Filed pursuant to Rule 424(b)(5)
Registration No. 333-235791
PROSPECTUS
SUPPLEMENT (To prospectus dated January 13, 2020)
$19,700,000
Common Stock
In
accordance with the terms of the Capital on DemandTM Sales Agreement
entered into with JonesTrading Institutional Services LLC
(“JonesTrading” or the “Agent”), dated
January 13, 2020, which we refer to as the sales agreement, we may
offer and sell under this prospectus supplement and accompanying
prospectus shares of our Common Stock, $0.001 par value per share,
having an aggregate offering price of up to $19,700,000 from time
to time through or to the Agent, acting as sales agent or
principal.
Our
Common Stock is listed for trading on the Nasdaq Capital Market
under the symbol “MNPR.” On January 10, 2020 the last
reported sale price of our Common Stock was $22.00 per
share.
The aggregate
market value of our outstanding Common Stock held by non-affiliates
was approximately $59.2 million based on 2,191,820 shares of
outstanding Common Stock held by non-affiliates and a price per
share of $27.00, the closing price of our Common Stock on December
20, 2019. Pursuant to General Instruction I.B.6 of Form S-3, in no
event will we sell securities pursuant to this prospectus
supplement with a value more than one-third of the aggregate market
value of our Common Stock held by non-affiliates in any 12-month
period, so long as the aggregate market value of our Common Stock
held by non-affiliates remains less than $75.0 million. In the
event that the aggregate market value of our outstanding Common
Stock held by non-affiliates equals or exceeds $75.0 million, then
the one-third limitation on sales may not apply to additional sales
made pursuant to this prospectus supplement. We have not sold any
securities pursuant to General Instruction I.B.6 of Form S-3 during
the 12 calendar months prior to, and including, the date of this
prospectus supplement.
Sales
of our Common Stock, if any, under this prospectus supplement will
be made by any method permitted that is deemed an “at the
market offering” as defined in Rule 415 under the Securities
Act of 1933, as amended (the “Securities Act”). The
Agent is not required to sell any specific amount of shares of our
Common Stock, but will act as our sales agent using commercially
reasonable efforts consistent with their normal trading and sales
practices. There is no arrangement for funds to be received in any
escrow, trust or similar arrangement.
The
Agent will be entitled to compensation at a commission rate of up
to 3.0% of the gross sales price per share of our Common Stock
sold. In connection with the sale of our Common Stock on our
behalf, the Agent will be deemed to be an “underwriter”
within the meaning of the Securities Act and the compensation of
the Agent will be deemed to be underwriting commission or discount.
We have also agreed to provide indemnification and contribution to
the Agent with respect to certain liabilities, including
liabilities under the Securities Act.
We are
an “emerging growth company” as that term is used in
the Jumpstart Our Business Startups Act of 2012 and, as such, have
elected to comply with certain reduced public company reporting
requirements for this prospectus supplement and future
filings.
Investing
in our Common Stock involves significant risks. See “Risk
Factors” beginning on page S-9 of this prospectus supplement
and in the documents incorporated by reference in this prospectus
supplement
for a discussion of the factors you should carefully consider
before deciding to purchase our Common Stock.
Neither the
Securities and Exchange Commission nor any state securities
commission has approved or disapproved of these securities or
passed upon the accuracy or adequacy of this prospectus
supplement.
Any representation to the contrary is a criminal
offense.
The
date of this Prospectus Supplement is January 13, 2020
TABLE
OF CONTENTS
|
Prospectus
Supplement
|
|
|
|
S-1
|
|
|
S-2
|
|
|
S-8
|
|
|
S-9
|
|
|
S-33
|
|
|
S-34
|
|
|
S-35
|
|
|
S-36
|
|
|
S-37
|
|
|
S-38
|
|
|
S-38
|
|
|
S-38
|
|
|
S-39
|
|
Prospectus
|
|
|
|
1
|
|
|
2
|
|
|
3
|
|
|
4
|
|
|
5
|
|
|
7
|
|
|
8
|
|
|
8
|
|
|
8
|
|
|
9
|
i
ABOUT THIS PROSPECTUS
SUPPLEMENT
This
prospectus supplement is a supplement to the accompanying
prospectus that is also a part of this document. This prospectus
supplement and the accompanying prospectus, dated January 13, 2020,
are part of a registration statement on Form S-3 (File No.
333-235791) that we filed with the Securities and Exchange
Commission (the “SEC”), utilizing a “shelf”
registration process. Under the shelf registration process, we may
offer and sell from time to time in one or more offerings our
Common Stock described in the accompanying prospectus.
This
document is in two parts. The first part is this prospectus
supplement, which describes our Common Stock we are offering and
the terms of the offering and also adds to and updates information
contained in the accompanying prospectus and the documents
incorporated by reference into the accompanying prospectus. The
second part is the accompanying prospectus, which provides more
general information. Generally, when we refer to this
“prospectus,” we are referring to both documents
combined. To the extent there is a conflict between the information
contained in this prospectus supplement, on the one hand, and the
information contained in the accompanying prospectus or any earlier
dated document incorporated by reference therein, on the other
hand, you should rely on the information in this prospectus
supplement. Additional prospectus supplements or documents filed
after the date hereof that are deemed incorporated by reference
herein may modify and supersede the information in this prospectus
supplement. We urge you to carefully read this prospectus
supplement and the accompanying prospectus and any related free
writing prospectus, together with the information incorporated
herein and therein by reference as described under the heading
“Incorporation of Information by Reference,” before
buying any of our Common Stock being offered.
You
should rely only on the information that we have provided or
incorporated by reference in this prospectus supplement and the
accompanying prospectus and any related free writing prospectus
that we may authorize to be provided to you. We have not, and Agent
has not, authorized anyone to provide you with different
information. No other dealer, salesperson or other person is
authorized to give any information or to represent anything not
contained in this prospectus supplement and the accompanying
prospectus or any related free writing prospectus that we may
authorize to be provided to you. You must not rely on any
unauthorized information or representation. This prospectus
supplement is an offer to sell only our Common Stock offered hereby
and only under circumstances and in jurisdictions where it is
lawful to do so. You should assume that the information in this
prospectus supplement and the accompanying prospectus or any
related free writing prospectus is accurate only as of the date on
the front of the document and that any information we have
incorporated by reference is accurate only as of the date of the
document incorporated by reference, regardless of the time of
delivery of this prospectus supplement and the accompanying
prospectus or any related free writing prospectus, or any sale of
our Common Stock.
This
prospectus supplement contains summaries of certain provisions
contained in some of the documents described herein, but reference
is made to the actual documents for complete information. All of
the summaries are qualified in their entirety by the actual
documents. Copies of some of the documents referred to herein have
been filed, will be filed or will be incorporated by reference as
exhibits to the registration statement of which this prospectus
supplement is a part, and you may obtain copies of those documents
as described below under the heading “Where You Can Find More
Information.”
As used
in this prospectus supplement, the terms “we”,
“us”, “our”, “Company”,
“Monopar Therapeutics” and “Monopar” refer
to Monopar Therapeutics Inc., a Delaware corporation.
We own
or have rights to trademarks or trade names that we use in
conjunction with the operation of our business. Each trademark,
trade name or service mark of any other company appearing in this
prospectus supplement or the accompanying prospectus belongs to its
holder. Use or display by us of other parties’ trademarks,
trade names or service marks is not intended to and does not imply
a relationship with, or endorsement or sponsorship by us of, the
trademark, trade name or service mark owner.
S-1
|
|
This summary highlights certain information about us, this offering
and selected information contained elsewhere in or incorporated by
reference into this prospectus. This summary is not complete and
does not contain all of the information that you should consider
before deciding whether to invest in our Common Stock. For a more
complete understanding of our Company and this offering, we
encourage you to read and consider carefully the more detailed
information in this prospectus, including the information
incorporated by reference into this prospectus, and the information
referred to under the heading “Risk Factors” in this
prospectus beginning on page S-9, and in the documents incorporated by
reference into this prospectus.
Overview
We are
a clinical stage biopharmaceutical company focused on developing
proprietary therapeutics designed to improve clinical outcomes for
cancer patients. We are building a drug development pipeline
through the licensing and acquisition of oncology therapeutics in
late preclinical and clinical development stages. We leverage our
scientific and clinical experience to help de-risk and accelerate
the clinical development of our drug product
candidates.
On
December 23, 2019, we completed our initial public offering. We
sold 1,277,778 shares of our Common Stock at a public offering
price of $8.00 per share. Net proceeds were approximately $9.3
million, after deducting underwriting discounts and offering
expenses. Our Common Stock began trading on the Nasdaq Capital
Market on December 19, 2019.
We are
devoting a significant portion of the net proceeds from our initial
public offering to prioritizing our
camsirubicin Phase 2 clinical trial which we recently signed a
collaboration agreement on, discussed in further detail
below. We are aiming to enroll the first patient in a Phase
3 clinical development program for our lead product candidate,
Validive (clonidine mucobuccal tablet; clonidine MBT) within a few
months of raising sufficient funds. To do so, we will require
additional funding in the millions or
tens of millions of dollars (depending on if we have a
collaboration or partnership or neither for Validive consummated),
or find a suitable pharmaceutical partner, both of which we are
planning to pursue in the coming months. There can be no
assurance that the funds raised from the sale of our Common Stock
pursuant to this prospectus supplement or other efforts will be
sufficient.
Validive is
designed to be used prophylactically to reduce the incidence, delay
the time to onset, and decrease the duration of severe oral
mucositis (“SOM”) in patients undergoing
chemoradiotherapy (“CRT”) for oropharyngeal cancer
(“OPC”). SOM is a painful and debilitating inflammation
and ulceration of the mucous membranes lining the oral cavity and
oropharynx in response to chemoradiation. The majority of patients
receiving CRT to treat their OPC develop SOM, which remains one of
the most common and devastating side effects of treatment in this
indication. The potential clinical benefits to patients of reducing
or delaying the incidence of SOM, or reducing the duration of SOM,
include: reduced treatment discontinuations leading to potentially
improved overall survival outcomes; reduced mouth and throat pain
avoiding the need to receive parenteral nutrition; and decreased
long-term and often permanent debilitation arising from swallowing
difficulties, neck and throat spasms, and lung complications due to
food aspiration. Our mucobuccal tablet (“MBT”)
formulation is a novel delivery system for clonidine that allows
for prolonged and enhanced local delivery of drug in the regions of
mucosal radiation damage in patients with OPC. Validive has been
granted fast track designation in the U.S., orphan drug designation
in the EU, and has global intellectual property patent protection
through mid-2029 not accounting for possible
extensions.
In
September 2017, we exercised an option to license Validive from
Onxeo S.A., the company that developed Validive through its Phase 2
clinical trial. In the completed Phase 2 clinical trial, Validive
demonstrated clinically meaningful efficacy signals within the
64-patient OPC population randomized to placebo, Validive 50
µg dose and Validive 100 µg dose. The absolute incidence
of SOM in OPC patients who received a dose of Validive 100 µg
once per day was reduced by 26.3% (incidence rate of 65.2% in
placebo, 45.0% in Validive 50 µg group, and 38.9% in Validive
100 µg group). The median time to onset of SOM was 37 days in
the placebo cohort; 45 days in the Validive 50 µg cohort and
no median time of onset was reached in the Validive 100 µg
group since fewer than half of this cohort of patients developed
SOM. There was also a 37.8% reduction in the median duration of the
SOM for the Validive 100 µg group versus placebo (41.0 days
placebo group, 34.0 days Validive 50 µg group, and 25.5 days
Validive 100 µg group) in patients that developed SOM. Median
duration of SOM across all patients, inclusive of both those that
did and did not develop SOM, was 17 days in the placebo group and 0
days in each of the Validive 50 and 100 µg groups. A positive
dose response was seen in each of these three clinical endpoints.
Additionally, patients in the Validive cohorts in the Phase 2
clinical trial demonstrated a safety profile similar to that of
placebo. While not designed by us, Onxeo’s promising
preclinical studies and Phase 2 clinical trial have informed the
design and conduct of what we believe will be an effective Phase 3
clinical program.
SOM
typically arises in the immune tissue at the back of the tongue and
throat, which comprise the oropharynx, and consists of acute severe
tissue damage and pain that prevents patients from swallowing,
eating and drinking. Validive stimulates the alpha-2 adrenergic
receptor on macrophages (white blood cells present in the immune
tissues of the oropharynx) suppressing pro-inflammatory cytokine
expression. Validive exerts its effects locally in the mouth over a
prolonged period of time through its unique MBT formulation.
Patients who develop SOM are also at increased risk of developing
late onset toxicities, including trismus (jaw, neck, and throat
spasms), dysphagia, and lung complications, which are often
irreversible and lead to increased hospitalization and the need for
further interventions sometimes years after completion of
chemoradiotherapy. We believe that a reduction in the incidence and
duration of SOM by Validive will have the potential to reduce
treatment discontinuation and/or treatment delays potentially
leading to improved survival outcomes, and reducing or eliminating
these long-term morbidities.
The OPC
target population for Validive is the most rapidly growing segment
of head and neck cancer (“HNC”) patients, with an
estimated 40,000 new cases of OPC in the alone in 2019. The growth
in OPC is driven by the increasing prevalence of oral human
papilloma virus (“HPV”) infections in the U.S. and
around the world. Despite the availability of a
pediatric/adolescent HPV vaccine, the rate of OPC incidence in
adults is not anticipated to be materially reduced for many decades
due to low adoption of the vaccine to date. As a result, the
incidence of HPV-driven OPC is projected to increase for many years
to come and will continue to support a clinical need for Validive
for the prevention of CRT-induced SOM in patients with OPC since
CRT is the standard of care treatment.
|
|
S-2
|
|
A
pre-Phase 3 meeting with the FDA was held and based on the meeting
discussion, a Phase 3 clinical protocol and accompanying
statistical analysis plan (“SAP”) was submitted to the
FDA for review and comments. We have also received protocol
assistance and advice on our Phase 3 protocol and SAP from the
European Medicines Agency Committee on Human Medicinal Products
(EMA/CHMP/SAWP). Based on comments and guidance provided by FDA and
EMA, subject to our ability to raise additional funding from the
sale of our Common Stock pursuant to this prospectus supplement, we
are aiming to enroll the first patient in our Phase 3 randomized
trial for our lead product candidate, Validive, within a few months
of raising sufficient funds. The Validive program will consist of
an adaptive design trial with an interim analysis planned for
approximately twelve months after the first patient is dosed, and a
confirmatory second trial planned to commence shortly after
completion of this interim analysis.
Our
second product candidate, camsirubicin, is a novel analog of
doxorubicin which has been designed to reduce the cardiotoxic side
effects generated by doxorubicin while retaining anti-cancer
activity. Camsirubicin is not metabolized to the derivatives that
are believed to be responsible for doxorubicin’s cardiotoxic
effects. A Phase 2 clinical trial for camsirubicin has been
completed in patients with advanced (e.g. unresectable or
metastatic) soft tissue sarcoma (“ASTS”). Average life
expectancy for these patients is 12-15 months. In this study, 52.6%
of patients evaluable for tumor progression demonstrated clinical
benefit (partial response or stable disease), which was
proportional to dose and consistently observed at higher cumulative
doses of camsirubicin (>1000 mg/m2). Camsirubicin was very well tolerated
in this study and underscored the ability to potentially administer
camsirubicin without restriction for cumulative dose in patients
with ASTS. Doxorubicin is limited to a lifetime cumulative dose
maximum of 450 mg/m2. Even if
a patient is responding, they are pulled off of doxorubicin
treatment once this cumulative dose has been reached.
Based
on encouraging clinical results to date, we plan to continue the
development of camsirubicin as first line treatment in patients
with ASTS, where the current first line treatment is doxorubicin.
The aim is to administer camsirubicin without restricting
cumulative dose, thereby potentially improving efficacy by keeping
patients on treatment who are responding. In June 2019, we entered
into a clinical collaboration with Grupo Español de
Investigación en Sarcomas (“GEIS”). GEIS will lead
a multi-country, randomized, open-label Phase 2 clinical trial
evaluating camsirubicin head-to-head against doxorubicin in
patients with ASTS. GEIS is an internationally renowned non-profit
organization focused on the research, development and management of
clinical trials for sarcoma that has worked with many of the
leading biotech and global pharmaceutical companies. Enrollment of
the trial is currently expected to begin in the first half of 2020
and to include approximately 170 ASTS patients, an interim
analysis, and take around two years to enroll. The primary endpoint
of the trial will be progression-free survival, with secondary
endpoints including overall survival and incidence of
treatment-emergent adverse events. In November 2019, the European
Commission granted orphan drug designation for camsirubicin for the
treatment of soft tissue sarcoma in the EU.
Our
third program, MNPR-101, is a novel first-in-class humanized
monoclonal antibody to the urokinase plasminogen activator receptor
(“uPAR”) for the treatment of advanced cancers. The
IND-enabling work is nearly completed.
Our
management team has extensive experience in developing therapeutics
through regulatory approval and commercialization. In aggregate,
companies they co-founded have achieved four drug approvals in the
U.S. and the EU, successfully sold an asset developed by management
which is currently in Phase 3 clinical trials, and completed the
sale of a biopharmaceutical company for over $800 million in cash.
Understanding the preclinical, clinical, regulatory and commercial
development processes and hurdles are key factors in successful
drug development and the expertise demonstrated by our management
team across all of these areas increases the probability of success
in advancing the product candidates in our product
pipeline.
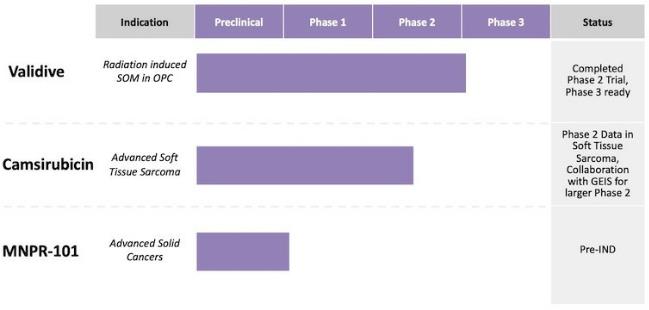
Our Product Pipeline
|
|
S-3
|
|
Our Product Candidates
Validive (clonidine mucobuccal tablet; clonidine MBT)
Validive is an MBT
of clonidine. The MBT formulation was developed to enhance the oral
mucosal drug delivery and significantly increase the salivary
concentrations of the active ingredient while minimizing systemic
absorption. The Validive tablet is tasteless and administered once
daily by affixing it to the outside of the patient’s upper
gum where it dissolves slowly over the period of several hours,
resulting in the extended release of clonidine into the oral cavity
and oropharynx, the site of SOM following chemoradiation treatment
for OPC. Validive therapy is designed to begin on the first day of
chemoradiation treatment and continue daily through the last day of
treatment.
SOM is
a painful and debilitating inflammation and ulceration of the
mucous membranes lining the oral cavity and oropharynx in response
to chemoradiation therapy. Patients receiving CRT to treat their
OPC often develop SOM, which remains one of the most common and
devastating side effects of treatment in this indication. We
believe Validive has the potential to address several critical
elements that affect SOM patients, including:
Reduction in the incidence of SOM. SOM
can increase the risk of acute and chronic comorbidities, including
dysphagia, trismus and lung complications, which are often
irreversible and lead to increased hospitalization and the need for
additional interventions. In a Phase 2 clinical trial, the OPC
patient cohort treated with Validive 100 µg demonstrated a
reduction in the absolute incidence of SOM compared to placebo of
26.3% (incidence rate of 65.2% in placebo, 45.0% in Validive 50
µg group, 38.9% in Validive 100 µg group). A reduced
incidence of SOM in OPC patients may lower the risk of acute and
chronic comorbidities and improve quality of life.
Delay in the time to onset of SOM. SOM
can cause cancer treatment delay and/or discontinuation, which may
impact overall survival outcomes. In a Phase 2 clinical trial, the
OPC patients had a time to onset of SOM of 37 days in the placebo
cohort; 45-day time to onset of SOM in the Validive 50 µg
cohort; and median was not reached as fewer than half of the
patients developed SOM in the Validive 100 µg group.
Prolonging time to onset of SOM may lead to fewer missed
chemoradiotherapy treatments, resulting in improved overall
survival outcomes.
Decrease in the duration of SOM. Longer
duration of SOM leads to a higher risk of the need for parenteral
nutrition and lower quality of life. SOM patients experience
inability to drink and/or eat, and difficulty swallowing often
resulting in malnourishment and feeding tube intervention. The
Phase 2 clinical trial data demonstrated a 15.5-day reduction (by
37.8%) in the duration of SOM for patients treated with Validive
100 µg (41 day median duration with placebo, 34 days with the
Validive 50 µg group, and 25.5 days for the Validive 100
µg group) in patients that developed SOM. Median duration
across all patients, inclusive of both those that did and did not
develop SOM, was 17 days in the placebo group and 0 days in each of
the Validive 50 and 100 µg groups. Reduced duration of SOM may
result in lower risk of malnourishment and feeding tube
intervention, and fewer treatment terminations/delays.
Camsirubicin (5-imino-13-deoxydoxorubicin; formerly MNPR-201,
GPX-150)
Camsirubicin is a
proprietary doxorubicin analog that is selective for topoisomerase
II-alpha. Doxorubicin is used to treat adult and pediatric solid
and blood (hematologic) cancers, including soft tissue sarcomas,
breast, gastric, ovarian and bladder cancers, leukemias and
lymphomas. The clinical efficacy of doxorubicin has historically
been limited by the risk of patients developing irreversible,
potentially life-threatening cardiotoxicity, despite clinical
studies demonstrating the anti-cancer benefit of higher doses of
doxorubicin administered for longer periods of time. For example,
several clinical studies completed in the 1990’s demonstrated
that concurrent doxorubicin (60 mg/m2, 8 cycles) and paclitaxel gave a 94%
overall response rate in patients with metastatic breast cancer but
led to 18% of these patients developing congestive heart failure.
Reduction of doxorubicin to 4-6 cycles of treatment decreased the
incidence of congestive heart failure, but also reduced response
rates to 45-55%.
Camsirubicin has
been engineered specifically to retain the anticancer activity of
doxorubicin while minimizing the toxic effects on the heart.
Similar to doxorubicin, the antitumor effects of camsirubicin are
mediated through the stabilization of the topoisomerase II complex
after a DNA strand break and DNA intercalation leading to tumor
cell apoptosis (cell death). Inhibiting the topoisomerase II-alpha
isoform is desired for the anti-cancer effect, while inhibiting the
topoisomerase II-beta isoform has been demonstrated to mediate, at
least in part, the cardiotoxicity associated with doxorubicin.
Camsirubicin is more selective than doxorubicin for inhibiting
topoisomerase II-alpha versus topoisomerase II-beta. This
selectivity may at least partly explain the minimal cardiotoxicity
that has been observed for camsirubicin in preclinical and clinical
studies to date. We believe these attributes provide a strong
rationale to develop camsirubicin without restriction on cumulative
dose, in a broad spectrum of cancer types.
|
|
S-4
|
|
Development of
camsirubicin is being pursued initially in patients with advanced
soft tissue sarcoma (ASTS). Currently, these patients receive
doxorubicin in the 1st line and
camsirubicin will be evaluated in a randomized Phase 2 trial head
to head against doxorubicin. Although doxorubicin has been the
standard of care treatment for over 40 years for patients with
ASTS, patients are pulled off treatment to limit irreversible heart
failure once the cumulative dose reaches 450 mg/m2, even if they are experiencing clinical
benefit. As a result, median progression free survival for ASTS
patients is approximately 6 months, with median overall survival of
12-15 months. Thus, there is a significant unmet opportunity to
develop a replacement for doxorubicin that retains anti-cancer
activity while reducing or eliminating the risk for irreversible
heart failure.
MNPR-101 (formerly huATN-658)
MNPR-101 is a
novel, preclinical stage drug candidate. It is a first-in-class
humanized monoclonal antibody to the urokinase plasminogen
activator receptor (“uPAR”), a well-credentialed cancer
therapeutic target. uPAR is a protein receptor that sits on the
cell surface of, and is overexpressed in, many deadly cancers, but
has little to no expression in healthy tissue; several Phase 1
imaging studies in human advanced cancer patients show that uPAR is
detected selectively in the tumor.
In
normal cells, uPAR is transiently expressed as part of a highly
regulated process required for the breakdown of the extracellular
matrix during normal tissue remodeling. In cancer, however, uPAR is
constitutively overexpressed by the tumor cell, and the uPAR
extracellular matrix degrading function is hijacked by the tumor to
support tissue invasion, metastasis, and angiogenesis. It is
important to tumor cell survival, and uPAR expression increases in
high grade and metastatic disease.
MNPR-101 has
demonstrated significant antitumor activity in numerous preclinical
models of tumor growth, both as a monotherapy and in combination
with other therapeutics and is being advanced toward an IND. Based
on the selective expression of uPAR in numerous tumor types, we
anticipate MNPR-101 will be well-tolerated and amenable to a
variety of combination treatment approaches in the
clinic.
Our Strategy
Leveraging the
experience and the demonstrated competencies of our management
team, our strategic goal is to acquire, develop and commercialize
promising oncology product candidates that address the unmet
medical needs of cancer patients. The five key elements of our
strategy to achieve this goal are to:
●
Advance the clinical development of
camsirubicin, by pursuing clinical indications where doxorubicin
has demonstrated efficacy. ASTS will be the first
indication, which will allow camsirubicin to go head-to-head
against doxorubicin, the current 1st line treatment. In
this indication, camsirubicin previously demonstrated clinical
benefit (stable disease or partial response) in 52.6% of patients
evaluable for tumor progression in a single arm Phase 2 study.
Clinical benefit was proportional to dose and consistently observed
at higher cumulative doses of camsirubicin (>1000
mg/m2). Camsirubicin was very
well tolerated in this Phase 2 study and underscored the ability to
potentially administer camsirubicin without restriction for
cumulative dose (doxorubicin is limited to 450 mg/m2 cumulative dose due to heart
toxicity).
●
Leverage data generated from the Phase 2
Validive clinical trial to position us well for a successful Phase
3 clinical program for Validive for SOM in OPC. In a Phase 2
clinical trial the absolute incidence of SOM in OPC patients was
reduced by 26.3%, the time to onset was delayed, and the duration
in patients that developed SOM was decreased by 15.5 days in the
Validive 100 µg cohort versus placebo. In addition to the data
from the Phase 2 clinical trial, we believe the guidance from our
key opinion leaders (“KOLs”) as well as from the FDA
and EMA, and our own internal clinical trial design expertise,
position us well for a successful Phase 3 clinical trial
program.
●
Obtain FDA approval of Validive and maximize
the commercial potential of Validive in the U.S. and the EU,
seeking partnerships outside these markets. Following a
potentially successful Phase 3 clinical program of Validive and
potential FDA approval, we currently intend to commercialize
Validive in the U.S. and the EU which may include establishing our
own specialty sales force and seeking partnerships outside of these
territories for regulatory approval and drug sales and
distribution.
●
Continue the development of MNPR-101 and expand
our drug development pipeline through in-license and acquisition of
oncology product candidates. We plan to continue the
development of MNPR-101 and the expansion of our drug development
pipeline through acquiring or in-licensing additional oncology
product candidates, particularly those that leverage existing
scientific and clinical data that helps de-risk the next steps in
clinical development.
●
Utilize the expertise and prior experience of
our team in the areas of asset acquisition, drug development and
commercialization to establish ourselves as a leading
biopharmaceutical company. Our senior executive team has
relevant experience in biopharmaceutical in-licensing and
acquisitions as well as developing product candidates through
approval and commercialization. In aggregate, our team has
co-founded BioMarin Pharmaceutical (Nasdaq: BMRN), Raptor
Pharmaceuticals ($800 million sale to Horizon Pharma), and Tactic
Pharma, LLC (“Tactic Pharma”) (sale of lead asset,
choline tetrathiomolybdate, which was ultimately acquired by
Alexion in June 2018 for $764 million).
|
|
S-5
|
|
Risks Associated with our Business
Our business is subject to numerous risks and uncertainties,
including those highlighted in the section titled
“Risk Factors” immediately following this prospectus
summary. These risks include, among others, the
following:
●
We are a clinical
stage biopharmaceutical company with a history of losses. We expect
to continue to incur significant losses for the foreseeable future
and may never achieve or maintain profitability, which could result
in a decline in the market value of our common stock.
●
Funds raised in our
recent initial public offering of our Common Stock are not
sufficient to start our Phase 3 clinical development of Validive,
and require that we raise significant additional funds from the
sale of our Common Stock pursuant to this prospectus supplement or
other financing efforts. If we are unable to raise enough funds in
the coming months from the sale of our Common Stock pursuant to
this prospectus supplement or other financing efforts, we may have
to consider strategic options such as out-licensing Validive,
entering into a clinical partnership, or terminating the program
there can be no assurance that we can find a suitable partner on
satisfactory terms.
●
We have a limited
operating history, no revenues from operations, and are dependent
upon raising capital to continue our drug development
programs.
●
We do not have and
may never have any approved products on the market. Our business is
highly dependent upon receiving approvals from various U.S. and
international governmental agencies and will be severely harmed if
we are not granted approval to manufacture and sell our product
candidates.
●
Our clinical trials
may not yield sufficiently conclusive results for regulatory
agencies to approve the use of our products.
●
If we experience
delays or difficulties in the enrollment of subjects in clinical
trials, our receipt of necessary regulatory approvals could be
delayed or prevented, which would materially affect our financial
condition.
●
We rely on third
parties to conduct our non-clinical studies and our clinical
trials. If these third parties do not successfully carry out their
contractual duties or meet expected deadlines, we may be unable to
obtain regulatory approval for or commercialize our current product
candidates or any future products and our financial condition will
be adversely affected.
●
We face significant
competition from other biotechnology and pharmaceutical companies,
and our operating results will suffer if we fail to compete
effectively. Competition and technological change may make our
product candidates obsolete or non-competitive.
●
The termination of
third-party licenses could adversely affect our rights to important
compounds or technologies.
●
If we and our
third-party licensors do not obtain and preserve protection for our
respective intellectual property rights, our competitors may be
able to take advantage of our development efforts to develop
competing drugs.
●
If we lose key
management leadership, and/or scientific personnel, and if we
cannot recruit qualified employees or other significant personnel,
we may experience program delays and increased compensation costs,
and our business may be materially disrupted.
Implications of Being an Emerging Growth Company
We
qualify as an “emerging growth company” as defined in
the Jumpstart our Business Startups Act of 2012 (“JOBS
Act”). An emerging growth company may take advantage of
specified reduced reporting and other burdens that are otherwise
applicable generally to public companies. These provisions include,
but are not limited to:
●
inclusion of only
two years, as compared to three years, of audited financial
statements in addition to any required unaudited interim financial
statements with correspondingly reduced “Management’s
discussion and analysis of financial condition and results of
operations” disclosures;
●
an exemption from
the auditor attestation requirement in the assessment of our
internal control over financial reporting pursuant to the
Sarbanes-Oxley Act of 2002 (“Sarbanes-Oxley
Act”);
●
an exemption from
compliance with any new requirements adopted by the Public Company
Accounting Oversight Board (“PCAOB”) requiring
mandatory audit firm rotation;
●
reduced disclosure
about executive compensation arrangements; and
●
an exemption from
the requirement to seek non-binding advisory votes on executive
compensation or golden parachute arrangements.
We may
take advantage of these provisions until we are no longer an
emerging growth company. We will remain an emerging growth company
until the earliest of (1) the last day of the year (a) following
the fifth anniversary of the completion of our initial public
offering, (b) in which we have total annual gross revenue of at
least $1.07 billion or (c) in which we are deemed to be a large
accelerated filer, which means the market value of our common stock
that is held by non-affiliates exceeds $700 million as of the prior
June 30th, and (2) the date on which we have issued more than $1.0
billion in non-convertible debt during the prior three-year
period.
We have
elected to take advantage of certain of the reduced disclosure
obligations in the registration statement, of which this prospectus
is a part, and may elect to take advantage of other reduced
reporting requirements in future filings. As a result, the
information that we provide to our stockholders may be different
than you might receive from other public reporting companies in
which you hold equity interests.
The
JOBS Act permits an emerging growth company such as us to take
advantage of an extended transition period to comply with new or
revised accounting standards applicable to public companies until
those standards would otherwise apply to private companies. We have
irrevocably elected to opt out of this provision and, as a result,
we will comply with new or revised accounting standards when they
are required to be adopted by public companies that are not
emerging growth companies. In addition, we are also a
“smaller reporting company” as defined in Rule 12b-2 of
the Exchange Act and have elected to take advantage of certain of
the scaled disclosure requirements available to smaller reporting
companies such as avoiding the extensive narrative disclosure
required of other reporting companies, particularly in the
description of executive compensation.
|
|
S-6
|
|
Corporate Information
We were
formed as a Delaware limited liability company in December 2014,
with the name Monopar Therapeutics, LLC. In December 2015, we
converted to a Delaware C corporation. Our principal executive
offices are located at 1000 Skokie Blvd, Suite 350, Wilmette, IL
60091. Our telephone number is (847) 388-0349. Our corporate
website is located at www.monopartx.com. Any information contained
in, or that can be accessed through our website, is not
incorporated by reference in this prospectus.
Trademark notice
We have
registered trademarks with the U.S. Patent and Trademark Office
(“USPTO”), for the following trademarks:
“Validive”, “Baxefyn”,
“Vidarys”, “Cotilix”, “Arvita”
and “Clonidol”. All other trademarks, service marks and
trade names in this prospectus are the property of their respective
owners. We have omitted the ® and ™ designations, as
applicable, for the trademarks used herein.
|
|
S-7
|
|
|
Common Stock Offered By
Us
|
|
Shares of our
Common Stock having an aggregate offering price up to
$19,700,000
|
|
Manner of Offering
|
|
“At the
market offering” that may be made from time to time on
Nasdaq through or to the
Agent, as sales agent or principal. See the section entitled
“Plan of Distribution” below.
|
|
Common Stock to be Outstanding After This
Offering(1)
|
|
Up to 11,483,087
shares, assuming sales price at a price of $22.00 per share, which
was the closing price of our Common
Stock on Nasdaq on January 11, 2020. The actual number of
shares
issued will vary depending on the sales price under this
offering.
|
|
Use of Proceeds
|
|
We intend to use
the net proceeds of this offering for our operations, including,
but not limited to, general corporate purposes, which may include
research and development expenditures, clinical trial expenditures,
manufacture and supply of product and working capital. See the
section entitled “Use
of Proceeds” below.
|
|
Nasdaq Capital Market
Symbol
|
|
MNPR
|
|
Risk Factors
|
|
See “Risk
Factors” beginning on page S-9 and the other information
included in, or incorporated by reference into, this prospectus for
a discussion of certain factors you should carefully consider
before deciding to invest in our Common
Stock.
|
(1)
The Common Stock
outstanding after the offering is based on approximately 10,587,632
shares of our Common Stock outstanding as of January 11, 2020 and
the sale of 895,455 shares of our Common Stock at an assumed
offering price of $22.00 per share, the last reported sale price of
our Common Stock on Nasdaq on January 11, 2020, and excludes the
following:
●
1,096,463 shares of
our Common Stock issuable upon the exercise of outstanding stock
options (weighted-average exercise price of $3.06; 758,685 shares
vested); and
●
485,104 shares of
our Common Stock reserved for issuance under our 2016 Stock
Incentive Plan.
Except
as otherwise indicated, all information in this prospectus
assumes:
●
All shares
referenced above and throughout this prospectus take into account a
70-for-1 stock split of our Common Stock effected in March
2017.
|
S-8
Investing in our Common Stock involves a high
degree of risk. Before deciding to invest in our Common Stock, you
should consider carefully the risks and uncertainties described
below, together with all of the other information in our Annual
Report on Form 10-K for the year ended December 31, 2018, including
the consolidated financial statements and the related notes, as
well as the risks and uncertainties in our subsequent quarterly
reports on Form 10-Q, as updated by our subsequent filings under
the Securities Exchange Act of 1934, as amended (the
“Exchange Act”) and other information set forth in our
reports filed with the SEC, all of which are incorporated by
reference into this prospectus in its entirety, together with other information in this
prospectus, and the information and documents incorporated by
reference in this prospectus, any prospectus supplement and any
free writing prospectus that we have authorized for use in
connection with this offering before you make a decision to invest
in our Common Stock. If any of the following risks or the risk
factors incorporated by reference hereto were to materialize, our
business, financial condition, results of operations, and future
growth prospects could be materially and adversely affected. In
that event, the market price of our Common Stock could decline, and
you could lose part of or all of your investment in our Common
Stock.
Risks Associated to our Common Stock and this Offering
Existing and new investors will experience
dilution as a result of future sales or issuances of our common
stock and future option exercises under our stock option
plan.
Our
Board Members, employees, and certain of our consultants have been
and will be issued equity and/or granted options that vest with the
passage of time. Up to a total of 1,600,000 shares of our common
stock may be issued as stock options or restricted stock under the
Amended and Restated Monopar Therapeutics Inc. 2016 Stock Incentive
Plan, and stock options for the purchase of up to 1,096,463 shares
of our common stock have already been granted (758,685 stock
options are exercisable) and are outstanding as of January 13,
2020. The issuance of such equity and/or the exercise of such
options will dilute both our existing and our new investors. As of
January 13, 2020, 18,433 stock options have been
exercised.
Our
existing and our new investors will likely also experience
substantial dilution resulting from the issuance by us of equity
securities in connection with certain transactions, including
without limitation, future offering of shares, intellectual
property licensing, acquisition, or commercialization
arrangements.
Holders of the shares of our
Common Stock will have no control of our operations or of decisions
on major transactions.
Our
business and affairs are managed by or under the direction of our
Board of Directors (“Board”). Our Stockholders are
entitled to vote only on actions that require a Stockholder vote
under federal or state law. Stockholder approval requires the
consent and approval of holders of a majority or more of our
outstanding stock. Shares of stock do not have cumulative voting
rights and therefore, holders of a majority of the shares of our
outstanding stock will be able to elect all Board Members.
TacticGem LLC (“TacticGem”) owns 7,166,667 shares of
common stock (68%). The limited liability company agreement of
TacticGem provides that the manager will vote its shares of Monopar
to elect to the Board those persons nominated by Tactic Pharma plus
one person nominated by Gem Pharmaceuticals, LLC
(“Gem”). Additionally, other than in the elections of
directors the limited liability company agreement requires
TacticGem to pass through votes to its members in proportion to
their membership percentages in TacticGem. As a result, Tactic
Pharma, our initial investor and participant in our initial public
offering, holds an approximately 42% beneficial interest in us and
together with Gem’s beneficial ownership of approximately
29%, the two entities control a majority of our stock and will be
able to elect all Board Members and control our affairs. Some of
our Board Members and executive officers own and control Tactic
Pharma. Although no single person has a controlling interest in
Tactic Pharma, acting together, they are able to control Tactic
Pharma and a large voting block of our Common Stock and elect over
a majority of our Board.
Our failure to meet the continued listing requirements of The
Nasdaq Capital Market could result in a de-listing of our Common
Stock.
If we
fail to satisfy the continued listing requirements of The Nasdaq
Capital Market, such as the corporate governance requirements or
the minimum closing bid price requirement, the Nasdaq Stock Market
(“Nasdaq”) may take steps to de-list our Common Stock.
Such a de-listing or the announcement of such de-listing will have
a negative effect on the price of our Common Stock and would impair
your ability to sell or purchase our Common Stock when you wish to
do so. In the event of a de-listing, we would take actions to
restore our compliance with the Nasdaq listing requirements, but we
can provide no assurance that any such action taken by us would
allow our Common Stock to become listed again, stabilize the market
price or improve the liquidity of our Common Stock, prevent our
Common Stock from dropping below the Nasdaq minimum bid price
requirement or prevent future non-compliance with the Nasdaq
listing requirements.
The stock price of our Common Stock may be volatile or may decline
regardless of our operating performance.
The
market prices for securities of biotechnology and pharmaceutical
companies have historically been highly volatile, and the market
has from time to time experienced significant price and volume
fluctuations that are unrelated to the operating performance of
particular companies. Our Common Stock has only been trading on the
Nasdaq Capital Market since December 19, 2019 and has experienced
significant volatility in market prices to date. Our small public
float and relatively low trading volumes exacerbate
volatility.
S-9
The
market price of our Common Stock is likely to remain highly
volatile and may fluctuate substantially due to many factors,
including:
●
announcements
concerning the progress and success of our clinical trials, our
ability to obtain regulatory approval for and commercialize our
product candidates, including any requests we receive from the FDA
for additional studies or data that result in delays in obtaining
regulatory approval or launching our product candidates, if
approved;
●
market conditions
in the pharmaceutical and biotechnology sectors or the economy as a
whole;
●
price and volume
fluctuations in the overall stock market;
●
the failure of our
product candidates, if approved, to achieve commercial
success;
●
announcements of
the clinical success, NDA approval or introduction of new products
by us or our competitors;
●
developments
concerning product development results or intellectual property
rights of others;
●
litigation or
public concern about the safety of our potential
products;
●
actual fluctuations
in our quarterly or annual operating results, and concerns by
investors that such fluctuations may occur in the future and are
symbolic of internal problems;
●
deviations in our
operating results from the estimates of securities analysts or
other analyst comments;
●
additions or
departures of key personnel;
●
healthcare reform
legislation, including measures directed at controlling the pricing
of pharmaceutical products, and third-party coverage and
reimbursement policies;
●
developments
concerning current or future strategic collaborations;
and
●
discussion of us,
our stock price or our potential future market value by the
financial and scientific press and in online investor
communities.
Investors in this Offering will suffer immediate and substantial
dilution of their investment.
The offering price per share
in this offering may exceed the net tangible book value per share
of our Common Stock. Assuming that an aggregate of 895,455 shares
of our Common Stock are sold at a price of $22.00 per share
pursuant to this prospectus, which was the last reported sale price
of our Common Stock on Nasdaq on January 11, 2020, for aggregate
net proceeds of $19,069,000 after deducting commissions and
estimated aggregate offering expenses payable by us, you would
experience immediate dilution of $19.12 per share, representing a
difference between our as adjusted net tangible book value per
share as of September 30, 2019 after giving effect, our recent
initial public offering, stock option exercises and this offering
and the assumed offering price. The exercise of outstanding stock
options and warrants may result in further dilution of your
investment. See the section entitled “Dilution” on page
S-38 of this prospectus for a more detailed illustration of the
dilution you would incur if you participate in this
offering.
We may become involved in securities class action litigation that
could divert management’s attention and harm our
business.
The
stock markets have from time to time experienced significant price
and volume fluctuations that have affected the market prices for
the common stock of biotechnology and pharmaceutical companies. Our
stock price has experienced such fluctuations since our initial
public offering. These broad market fluctuations may cause the
market price of our stock to decline. In the past, securities class
action litigation has often been brought against a company
following a decline in the market price of its securities. This
risk is especially relevant for us because biotechnology and
biopharmaceutical companies have experienced significant stock
price volatility in recent years. We may become involved in this
type of litigation in the future. Litigation often is expensive and
diverts management’s attention and resources, which could
adversely affect our business.
Substantial amounts of our outstanding shares may be sold into the
market when lock-up or market standoff periods end. If there are
substantial sales of shares of our Common Stock, the price of our
Common Stock could decline.
The
price of our Common Stock could decline if there are substantial
sales of our Common Stock, particularly sales by our directors,
executive officers and significant stockholders, or if there is a
large number of shares of our Common Stock available for sale and
the market perceives that sales will occur. We have 10,587,632
outstanding shares of our Common Stock as of the date of this
prospectus supplement. The overwhelming majority of all of our
outstanding shares of Common Stock are currently restricted from
resale as a result of market standoff and “lock-up”
agreements. These shares will become available to be sold after
June 16, 2020. Shares held by directors, executive officers and
other affiliates will be subject to volume limitations under Rule
144 under the Securities Act of 1933, as amended (Securities Act),
and various vesting agreements.
Certain
of our stockholders will have rights, subject to some conditions,
to require us to file registration statements covering their shares
or to include their shares in registration statements that we may
file for ourselves or our stockholders, subject to market standoff
and lock-up agreements. We have also registered shares of Common
Stock that we have issued and may issue under our employee equity
incentive plans. These shares are able to be sold freely in the
public market upon issuance, subject to existing market standoff or
lock-up agreements or internal practices which prohibit sales under
certain circumstances. The market price of the shares of our Common
Stock could decline as a result of the sale of a substantial number
of our shares of Common Stock in the public market or the
perception in the market that the holders of a large number of
shares intend to sell their shares.
S-10
We have broad discretion in the use of the net proceeds from this
offering, and our use of those proceeds may not yield a favorable
return on your investment.
We
intend to use the net proceeds of this offering for our operations,
including, but not limited to, general corporate purposes, which
may include research and development expenditures, clinical trial
expenditures, manufacture and supply of product and working
capital, We have not specifically allocated the amount of net
proceeds that will be used for these purposes, and our management
will have broad discretion over how these proceeds are used and
could spend the proceeds in ways with which you may not agree. In
addition, we may not use the proceeds of this offering effectively
or in a manner that increases our market value or enhances our
profitability. We have not established a timetable for the
effective deployment of the proceeds, and we cannot predict how
long it will take to deploy the proceeds.
Our ability to use our net operating loss carry-forwards and
certain other tax attributes may be limited.
Under
Section 382 of the Code, if a corporation undergoes an
“ownership change” (generally defined as a greater than
50% change, by value, in its equity ownership over a three-year
period), the corporation’s ability to use its pre-change net
operating loss carry-forwards and other pre-change tax attributes
(such as research tax credits) to offset its post-change income may
be limited. We believe that additional fundraising efforts in the
next three years, may trigger an “ownership change”
limitation in the near future. As a result, if we earn net taxable
income, our ability to use our pre-change net operating loss
carry-forwards to offset U.S. federal taxable income will be
subject to limitations, which could result in increased future tax
liability to us had we not been subject to such
limitations.
If securities or industry analysts do not publish research or
publish inaccurate or unfavorable research about our business, our
stock price and trading volume could decline.
The
trading market for our common stock will depend in part on the
research and reports that securities or industry analysts publish
about us or our business. Securities and industry analysts do not
currently, and may never, publish research on our Company. If
securities or industry analysts do not commence coverage of our
Company, the trading price for our stock would likely be negatively
impacted. In the event securities or industry analysts initiate
coverage, if one or more of the analysts who covers us downgrades
our stock or publishes inaccurate or unfavorable research about our
business, our stock price may decline. If one or more of these
analysts ceases coverage of our Company or fails to publish reports
on us regularly, demand for our stock could decrease, which might
cause our stock price and trading volume to decline.
We do not intend to pay dividends for the foreseeable future and,
as a result, your ability to achieve a return on your investment
will depend on appreciation in the price of our Common
Stock.
We have
never declared or paid any cash dividends on our capital stock and
we do not intend to pay any cash dividends in the foreseeable
future. Any determination to pay dividends in the future will be at
the discretion of our Board. Accordingly, investors must rely on
sales of their common stock after price appreciation, which may
never occur, as the only way to realize any future gains as a
return on their investments.
There can be no assurance that we will ever provide liquidity to
our investors through a sale of our Company.
While
acquisitions of pharmaceutical companies like ours are not
uncommon, potential investors are cautioned that no assurances can
be given that any form of merger, combination, or sale of our
Company will take place or that any merger, combination, or sale,
even if consummated, would provide liquidity or a profit for our
investors. You should not invest in our Company with the
expectation that we will be able to sell the business in order to
provide liquidity or a profit for our investors.
Delaware law and provisions in our amended and restated bylaws
could make a merger, tender offer or proxy contest difficult,
thereby depressing the potential trading price of our Common
Stock.
Anti-takeover
provisions in our charter documents and under Delaware law could
make an acquisition of us difficult, limit attempts by our
stockholders to replace or remove our current management or Board
and adversely affect our stock price.
Provisions of our
amended and restated bylaws may delay or discourage transactions
involving an actual or potential change in our control or change in
our management, including transactions in which stockholders might
otherwise receive a premium for their shares, or transactions that
our stockholders might otherwise deem to be in their best
interests. Therefore, these provisions could adversely affect the
price of our stock. Among other things, our amended and restated
bylaws:
●
provide that all
vacancies on our Board may only be filled by our Board and not by
stockholders;
●
allow
the holders of a plurality of the shares of common stock entitled
to vote in any election of directors to elect all of the directors
standing for election, if they should so choose; and
●
provide that
special meetings of our stockholders may be called only by our
Board.
In
addition, because we are incorporated in Delaware, we are governed
by the provisions of Section 203 of the Delaware General
Corporation Law, which generally prohibits a Delaware corporation
from engaging in any of a broad range of business combinations with
any “interested” stockholder for a period of three
years following the date on which the stockholder became an
“interested” stockholder.
S-11
Risks Related to Our Financial Condition and Capital
Requirements
We have a limited operating history, expect to incur significant
operating losses, and have a high risk of never being
profitable.
We
commenced operations in December 2014 and have a limited operating
history of five years. Therefore, there is limited historical
financial or operational information upon which to evaluate our
performance. Our prospects must be considered in light of the
uncertainties, risks, expenses, and difficulties frequently
encountered by companies in their early stages of operations. Many
if not most companies in our industry at our stage of development
never become profitable and are acquired or go out of business
before successfully developing any product that generates revenue
from commercial sales or enables profitability.
From
inception in December 2014 through September 30, 2019, we have
incurred losses of approximately $24.7 million, which includes
$13.5 million of non-cash in-process research and development,
which was incurred in connection with our acquisition of
camsirubicin. We expect to continue to incur substantial operating
losses over the next several years for the clinical development of
our current and future licensed or purchased product
candidates.
The
amount of future losses and when, if ever, we will become
profitable are uncertain. We do not have any products that have
generated any revenues from commercial sales, and do not expect to
generate revenues from the commercial sale of products in the near
future, if ever. Our ability to generate revenue and achieve
profitability will depend on, among other things, successful
completion of the development of our product candidates; obtaining
necessary regulatory approvals from the FDA and international
regulatory agencies; establishing manufacturing, sales, and
marketing arrangements with third parties; obtaining adequate
reimbursement by third-party payers; and raising sufficient funds
to finance our activities. If we are unsuccessful at some or all of
these undertakings, our business, financial condition, and results
of operations are expected to be materially and adversely
affected.
As a
recently established public reporting company, we are subject to
SEC reporting and other requirements, which will lead to increased
operating costs in order to meet these requirements.
The funds raised from the recent initial public offering of our
Common Stock should enable us to obtain topline results for the
camsirubicin Phase 2 clinical trial and ramp up the initiation of
Validive’s Phase 3 clinical program, but are not sufficient
for us to start our Validive Phase 3 clinical program, which
require that we raise significant additional funds from the sale of
our Common Stock pursuant to this prospectus supplement or other
financing efforts. If we are able to raise additional funds from
the sale of our Common Stock pursuant to this prospectus
supplement, to start our Phase 3 clinical trial for Validive, it
may not be on favorable terms. If we are unable to raise enough
funds in the future, we may have to discontinue or delay our
operations.
In
order to be commercially viable, we must successfully research,
develop, obtain regulatory approval for, manufacture, introduce,
market and distribute camsirubicin, Validive and, if applicable,
any current and future product candidates we may develop. The
estimated required capital and time-frames necessary to achieve
these developmental milestones as described in this prospectus or
as we may state from time to time is subject to inherent risks,
many of which may be beyond our control. Clinical development of
camsirubicin and Validive will require significant funds. Proceeds
from the recent initial public offering of our Common Stock should
enable us to obtain topline results for the camsirubicin Phase 2
clinical trial and ramp up the initiation of Validive’s Phase
3 clinical program, but are not sufficient for us to start our
Validive Phase 3 clinical program, which require that we raise
significant additional funds from the sale of our Common Stock
pursuant to this prospectus supplement or other financing efforts.
If we are unable to raise enough funds in the future, we may have
to discontinue or delay our operations.
If we continue to incur operating losses and fail to obtain the
capital necessary to fund our operations, we will be unable to
advance our development programs, complete our clinical trials, or
bring products to market, or may be forced to reduce or cease
operations entirely. In addition, any capital obtained by us may be
obtained on terms that are unfavorable to us, our investors, or
both.
Developing a new
drug and conducting clinical trials and the regulatory review
processes for one or more disease indications involves substantial
costs. We have projected cash requirements for the near term based
on a variety of assumptions, but some or all of such assumptions
are likely to be incorrect and/or incomplete, possibly materially
in an adverse direction. Our actual cash needs may deviate
materially from those projections, changes in market conditions or
other factors may increase our cash requirements, or we may not be
successful even in raising the amount of cash we currently project
will be required for the near term. We will need to raise
additional capital in the future; the amount of additional capital
needed will vary as a result of a number of factors, including
without limitation the following:
●
receiving less
funding than we require;
●
higher than
expected costs to manufacture our active pharmaceutical ingredient
and our product candidates;
●
higher than
expected costs for preclinical testing;
●
an increase in the
number, size, duration, and/or complexity of our clinical
trials;
●
slower than
expected progress in developing Validive, camsirubicin, MNPR-101,
or other product candidates, including without limitation,
additional costs caused by program delays;
●
higher than
expected costs associated with attempting to obtain regulatory
approvals, including without limitation additional costs caused by
additional regulatory requirements or larger clinical trial
requirements;
●
higher than
expected personnel, consulting or other costs, such as adding
personnel or industry expert consultants or pursuing the
licensing/acquisition of additional assets; and
●
higher than
expected costs to protect our intellectual property portfolio or
otherwise pursue our intellectual property strategy.
S-12
When we
attempt to raise additional financing, there can be no assurance
that we will be able to secure such additional financing in
sufficient quantities or at all. We may be unable to raise
additional capital for reasons including, without limitation, our
operational and/or financial performance, investor confidence in us
and the biopharmaceutical industry, credit availability from banks
and other financial institutions, the status of current projects,
and our prospects for obtaining any necessary regulatory approvals.
Potential investors’ capital investments may have shifted to
other opportunities with perceived greater returns and/or lower
risk thereby reducing capital available to us, if available at
all.
In
addition, any additional financing might not be available, and even
if available, may not be available on terms favorable to us or our
then-existing investors. We will seek to raise funds through public
or private equity offerings, debt financings, corporate
collaboration or licensing arrangements, mergers, acquisitions,
sales of intellectual property, or other financing vehicles or
arrangements. To the extent that we raise additional capital by
issuing equity securities or other securities, our then-existing
investors will experience dilution. If we raise funds through debt
financings or bank loans, we may become subject to restrictive
covenants, our assets may be pledged as collateral for the debt,
and the interests of our then-existing investors would be
subordinated to the debt holders or banks. In addition, our use of
and ability to exploit assets pledged as collateral for debt or
loans may be restricted or forfeited. To the extent that we raise
additional funds through collaboration or licensing arrangements,
we may be required to relinquish significant rights (including
without limitation intellectual property rights) to our
technologies or product candidates, or grant licenses on terms that
are not favorable to us. If we are not able to raise needed funding
under acceptable terms or at all, then we will have to reduce
expenses, including the possible options of curtailing operations,
abandoning opportunities, licensing or selling off assets, reducing
costs to a point where clinical development or other progress is
impaired, or ceasing operations entirely.
Unstable market and economic conditions may have serious adverse
consequences on our ability to raise funds, which may cause us to
cease or delay our operations.
From
time to time, global and domestic credit and financial markets have
experienced extreme disruptions, including severely diminished
liquidity and credit availability, declines in consumer confidence,
declines in economic growth, increases in unemployment rates, and
uncertainty about economic stability. Our financing strategy will
be adversely affected by any such economic downturn, volatile
business environment and continued unpredictable and unstable
market conditions. If the equity and credit markets deteriorate, it
may make a debt or equity financing more difficult to complete,
costlier, and more dilutive. Failure to secure any necessary
financing in a timely manner and on favorable terms will have a
material adverse effect on our business strategy and financial
performance, and could require us to cease or delay our
operations.
Risks Related to Clinical Development and Regulatory
Approval
We do not have and may never have any approved products on the
market. Our business is highly dependent upon receiving approvals
from various U.S. and international governmental agencies and will
be severely harmed if we are not granted approval to manufacture
and sell our product candidates.
In
order for us to commercialize any treatment for
chemoradiation-induced SOM or for any other disease indication, we
must obtain regulatory approvals of such treatment for that
indication. Satisfying regulatory requirements is an expensive
process that typically takes many years and involves compliance
with requirements covering research and development, testing,
manufacturing, quality control, labeling, and promotion of drugs
for human use. To obtain necessary regulatory approvals, we must,
among other requirements, complete clinical trials demonstrating
that our products are safe and effective for a particular
indication. There can be no assurance that our products will prove
to be safe and effective, that our clinical trials will demonstrate
the necessary safety and effectiveness of our product candidates,
or that we will succeed in obtaining regulatory approval for any
treatment we develop even if such safety and effectiveness are
demonstrated.
Any
delays or difficulties we encounter in our clinical trials may
delay or preclude regulatory approval from the FDA or from
international regulatory organizations. Any delay or preclusion of
regulatory approval would be expected to delay or preclude the
commercialization of our products. Examples of delays or
difficulties that we may encounter in our clinical trials include,
without limitation, the following:
●
Clinical trials may
not yield sufficiently conclusive results for regulatory agencies
to approve the use of our products.
●
Our products may
fail to be more effective than current therapies, or to be
effective at all.
●
We may discover
that our products have adverse side effects, which could cause our
products to be delayed or precluded from receiving regulatory
approval or otherwise expose us to significant commercial and legal
risks.
●
It may take longer
than expected to determine whether or not a treatment is safe and
effective.
●
Patients involved
in our clinical trials may suffer severe adverse side effects, even
up to death, whether as a result of treatment with our products,
the withholding of such treatment, or other reasons (whether within
or outside of our control).
●
We may fail to be
able to enroll a sufficient number of patients in our clinical
trials.
●
Patients enrolled
in our clinical trials may not have the characteristics necessary
to obtain regulatory approval for a particular indication or
patient population.
●
We may be unable to
produce sufficient quantities of product to complete the clinical
trials.
●
Even if we are
successful in our clinical trials, any required governmental
approvals may still not be obtained or, if obtained, may not be
maintained.
●
If approval for
commercialization is granted, it is possible the authorized use
will be more limited than is necessary for commercial success, or
that approval may be conditioned on completion of further clinical
trials or other activities, which will cause a substantial increase
in costs and which we might not succeed in performing or
completing.
●
If granted,
approval may be withdrawn or limited if problems with our products
emerge or are suggested by the data arising from their use or if
there is a change in law or regulation.
S-13
Any
success we may achieve at a given stage of our clinical trials does
not guarantee that we will achieve success at any subsequent stage,
including without limitation final FDA or other regulatory
organizations approval.
We may
encounter delays or rejections in the regulatory approval process
because of additional government regulation resulting from future
legislation or administrative action, or from changes in the
policies of the FDA or other regulatory bodies during the period of
product development, clinical trials, or regulatory review. Failure
to comply with applicable regulatory requirements may result in
criminal prosecution, civil penalties, recall or seizure of
products, total or partial suspension of production, or an
injunction preventing certain activity, as well as other regulatory
action against our product candidates or us. As a company, we have
no experience in successfully obtaining regulatory approval for a
product and thus may be poorly equipped to gauge, and may prove
unable to manage, risks relating to obtaining such
approval.
Outside
the U.S., our ability to market a product is contingent upon
receiving clearances from appropriate non-U.S. regulatory
authorities. Non-U.S. regulatory approval typically includes all of
the risks associated with FDA clearance discussed above as well as
geopolitical uncertainties and the additional uncertainties and
potential prejudices faced by U.S. pharmaceutical companies
conducting business abroad. In certain cases, pricing restrictions
and practices can make achieving even limited profitability very
difficult.
Even if we complete the clinical trials we discussed with the FDA,
there is no guarantee that at the time of submission the FDA will
accept our new drug application (“NDA”).
The FDA
provided helpful guidance on our proposed Validive adaptive design
trial and confirmatory second trial, informing us it might be an
acceptable pathway for NDA submission, but the FDA is not bound by
the guidance they give, and can change their position in the
future. Any future decision by the FDA will be driven largely by
the data generated from the Validive clinical trials.
As a company, we have never completed a clinical trial and have
limited experience in completing regulatory filings and any delays
in regulatory filings could materially affect our financial
condition.
While
members of our team have conducted numerous clinical trials at
previous companies, and have launched and marketed innovative
pharmaceutical products in the US and internationally, as a
company, we have not yet completed any clinical trials of our
product candidates, nor have we demonstrated the ability to obtain
marketing approvals, manufacture product candidates at a commercial
scale, or conduct sales and marketing activities necessary for the
successful commercialization of a product. Consequently, we have no
historical basis as a company by which one can evaluate or predict
reliably our future success or viability.
Additionally, while
our team has experience at prior companies with regulatory filings,
as a company, we have limited experience with regulatory filings
with agencies such as the FDA or EMA. Any delay in our regulatory
filings for our product candidates, and any adverse development or
perceived adverse development with respect to the applicable
regulatory authority’s review of such filings, including,
without limitation, the FDA’s issuance of a “refuse to
file” letter or a request for additional information, could
materially affect our financial condition.
We may seek fast track designation for one or more of our current
and future product candidates, but we might not receive such
designation, and even if we do, such designation may not actually
lead to a faster development or regulatory review or approval
process.
Our
lead product candidate, Validive, has been given fast track
designation from the FDA. Fast track designation does not ensure
that we will receive marketing approval or that approval will be
granted within any particular timeframe. We may not experience a
faster development, regulatory review or approval process with fast
track designation compared to conventional FDA procedures.
Additionally, the FDA may withdraw fast track designation, for
reasons such as it comes to believe a drug candidate no longer
adequately addresses an unmet medical need. Fast track designation
alone does not guarantee qualification for the FDA’s priority
review procedures. If we seek fast track designation for other
product candidates, we may not receive such a designation from the
FDA.
We, or any future collaborators, may not be able to obtain and
maintain orphan drug exclusivity for our product candidates in the
U.S. and Europe.
Validive has been
granted orphan drug designation for the treatment of SOM in the EU.
Camsirubicin has been granted orphan drug designation for the
treatment of soft tissue sarcoma in the U.S. and in the EU. We may
seek additional orphan drug designations or regulatory incentives
for our pipeline product candidates, for other indications or for
future product candidates. There can be no assurances that we will
be able to obtain such designations.
Even if
we obtain orphan drug designation for a product candidate, we may
not be able to maintain orphan drug exclusivity for that drug. For
example, orphan drug designation may be removed if the prevalence
of an indication increases beyond the patient number limit required
to maintain designation. Generally, if a drug with an orphan drug
designation subsequently receives the first marketing approval for
the indication for which it has such designation, the drug is
entitled to a period of marketing exclusivity, which precludes the
EMA or the FDA from approving another marketing application for the
same product in the same indication for that time period. Orphan
drug exclusivity may be lost if the FDA or EMA determines that the
request for designation was materially defective or if the
manufacturer is unable to assure sufficient quantity of the product
to meet the needs of patients with the rare disease or condition.
Moreover, even after an orphan drug is approved, the FDA can
subsequently approve a different drug for the same condition if the
FDA concludes that the later drug is clinically superior in that it
is shown to be safer, more effective or makes a major contribution
to patient care compared to our product.
The FDA
may reevaluate the Orphan Drug Act and its regulations and
policies, and similarly the EMA may reevaluate its policies and
regulations. We do not know if, when, or how the FDA or EMA may
change their orphan drug regulations and policies in the future,
and it is uncertain how any changes might affect our business.
Depending on what changes the FDA and/or EMA may make to their
orphan drug regulations and policies, our business could be
adversely impacted.
S-14
If serious adverse or undesirable side effects are identified
during the development of our product candidates, we may abandon or
limit our development or commercialization of such product
candidates.
If our
product candidates are associated with undesirable side effects or
have unexpected characteristics, we may need to abandon their
development or limit development to certain uses or subpopulations
in which the undesirable side effects or other characteristics are
less prevalent, less severe or more acceptable from a risk-benefit
perspective.
If we
elect or are forced to suspend or terminate any clinical trial with
one of our product candidates, the commercial prospects of such
product candidate will be harmed, and our ability to generate
revenue from such product candidate will be delayed or eliminated.
Any of these occurrences may harm our business, financial condition
and prospects significantly.
With
regard to our lead product candidate, Validive, unforeseen side
effects from Validive could arise either during clinical
development or, if approved, after Validive has been marketed. This
could cause regulatory approvals for, or market acceptance of,
Validive harder and costlier to obtain.
To
date, no difference in the frequency of serious adverse events
(“SAEs”) has been observed in patients treated with
Validive compared to placebo. In the Phase 2 clinical trial, two
patients in the placebo group and 2 patients in the Validive 50
µg group experienced SAEs that were assessed as treatment
related. No patients in the Validive treated cohorts were
discontinued due to study drug. Clonidine, the active ingredient of
Validive, has been used for over 50 years as an orally swallowed
systemic treatment for high blood pressure. Validive administration
leads to very low, but still detectable exposure of clonidine
outside the oral cavity. Thus, there is some risk that patients may
experience side effects due to this systemic exposure, which could
include a reduction in blood pressure, irregular heartbeat,
drowsiness or dry mouth.
The
results of our planned or any future clinical trials may show that
the side effects of Validive are unacceptable or intolerable, which
could interrupt, delay or halt clinical trials, and result in delay
of, or failure to obtain, marketing approval from the FDA or EMA
and other regulatory authorities, or result in marketing approval
from the FDA or EMA and other regulatory authorities with
restrictive label warnings.
If
Validive receives marketing approval and we or others later
identify undesirable or unacceptable side effects caused by the use
of Validive:
●
regulatory
authorities may withdraw their approval of the product, which would
force us to remove Validive from the market;
●
regulatory
authorities may require the addition of labeling statements,
specific warnings, a contraindication, or field alerts to
physicians and pharmacies;
●
we may be required
to change instructions regarding the way the product is
administered, conduct additional clinical trials or change the
labeling of the product;
●
we may be subject
to limitations on how we may promote the product;
●
sales of the
product may decrease significantly;
●
we may be subject
to litigation or product liability claims; and
●
our reputation may
suffer.
Any of
these events could prevent us or our potential future collaborators
from achieving or maintaining market acceptance of Validive and/or
could substantially increase commercialization costs and expenses,
which in turn could delay or prevent us from generating significant
revenues from the sale of Validive.
Our Phase 3 development program for Validive entails significant
risk.
The
Phase 3 development program for Validive has been designed based on
an analysis of the 64 oropharyngeal cancer (“OPC”)
patients included in the Phase 2 trial (n= 24 in the placebo group,
n= 21 Validive 50 µg group, and n= 19 Validive 100 µg
group). While a dose response was observed in the Validive treated
OPC cohorts compared to placebo across multiple clinically
meaningful endpoints, the ability to establish statistical
significance was limited by the relatively small sample size. This
increases the risk of the Phase 3 trials. Given the large unmet
medical need for the prevention of radiotherapy-induced SOM in OPC
patients, we have decided to pursue an adaptive design Phase 3
clinical development strategy in an effort to mitigate this risk.
Our adaptive design approach will allow us to confirm or reject our
hypothesis based off the Phase 2 data that the optimal patient
population for Validive is likely either all OPC patients or HPV+
OPC patients, and then run a confirmatory second trial should it be
warranted.
If we experience delays or difficulties in the enrollment of
subjects to our clinical trials, our receipt of necessary
regulatory approvals could be delayed or prevented, which could
materially affect our financial condition.
Identifying,
screening and enrolling patients to participate in clinical trials
of our product candidates is critical to our success, and we may
not be able to identify, recruit, enroll and dose a sufficient
number of patients with the required or desired characteristics to
complete our clinical trials in a timely manner. The timing of our
clinical trials depends on our ability to recruit patients to
participate as well as to subsequently dose these patients and
complete required follow-up periods. In particular, because our
planned clinical trials of Validive and camsirubicin are focused on
indications with relatively small patient populations, our ability
to enroll eligible patients may be limited or may result in slower
enrollment than we anticipate.
S-15
In
addition, we may experience enrollment delays related to increased
or unforeseen regulatory, legal and logistical requirements at
certain clinical trial sites. These delays could be caused by
reviews by regulatory authorities and contractual discussions with
individual clinical trial sites. Any delays in enrolling and/or
dosing patients in our planned clinical trials could result in
increased costs, delays in advancing our product candidates, delays
in testing the effectiveness of our product candidates or in
termination of the clinical trials altogether.
Patient
enrollment may be affected if our competitors have ongoing clinical
trials with products for the same indications as our product
candidates, and patients who would otherwise be eligible for our
clinical trials instead enroll in our competitors’ clinical
trials. Patient enrollment may also be affected by other factors,
including:
●
coordination with
clinical research organizations to enroll and administer the
clinical trials;
●
coordination and
recruitment of collaborators and investigators at individual
sites;
●
size of the patient
population and process for identifying patients;
●
design of the
clinical trial protocol;
●
eligibility and
exclusion criteria;
●
perceived risks and
benefits of the product candidates under study;
●
availability of
competing commercially available therapies and other competing
products’ clinical trials;
●
time of year in
which the trials are initiated or conducted;
●
severity of the
diseases under investigation;
●
ability to obtain
and maintain subject consents;
●
ability to enroll
and treat patients in a timely manner;
●
risk that enrolled
subjects will drop out before completion of the
trials;
●
proximity and
availability of clinical trial sites for prospective
patients;
●
ability to monitor
subjects adequately during and after treatment; and
●
patient referral
practices of physicians.
Our
inability to enroll a sufficient number of patients for clinical
trials would result in significant delays and could require us to
abandon one or more clinical trials altogether. Enrollment delays
in these clinical trials may result in increased development costs
for our product candidates, which could materially affect our
financial condition.
If we or our licensees, development collaborators, or suppliers are
unable to manufacture our products in sufficient quantities or at
defined quality specifications, or are unable to obtain regulatory
approvals for the manufacturing facility, we may be unable to
develop and/or meet demand for our products and lose time to market
and potential revenues.
Completion of our
clinical trials and commercialization of our product candidates
require access to, or development of, facilities to manufacture a
sufficient supply of our product candidates. We will utilize third
parties to manufacture Validive, camsirubicin, and MNPR-101. We
currently have manufacturing agreements in place for Validive and
MNPR-101. We are in negotiations with manufacturers for
camsirubicin.
In the
future we may become unable, for various reasons, to rely on our
sources for the manufacture of our product candidates, either for
clinical trials or, at some future date, for commercial
distribution. We may not be successful in identifying additional or
replacement third-party manufacturers, or in negotiating acceptable
terms with any we do identify. We may face competition for access
to these manufacturers’ facilities and may be subject to
manufacturing delays if the manufacturers give other clients higher
priority than they give to us. Even if we are able to identify an
additional or replacement third-party manufacturer, the delays and
costs associated with establishing and maintaining a relationship
with such manufacturer may have a material adverse effect on
us.
Before
we can begin to commercially manufacture Validive, camsirubicin,
MNPR-101, or any other product candidate, we must obtain regulatory
approval of the manufacturing facility and process. Manufacturing
of drugs for clinical and commercial purposes must comply with
current Good Manufacturing Practices requirements, commonly known
as “cGMP.” The cGMP requirements govern quality control
and documentation policies and procedures. Complying with cGMP and
non-U.S. regulatory requirements will require that we expend time,
money, and effort in production, recordkeeping, and quality control
to ensure that the product meets applicable specifications and
other requirements. We, or our contracted manufacturing facility,
must also pass a pre-approval inspection prior to FDA approval.
Failure to pass a pre-approval inspection may significantly delay
or prevent FDA approval of our products. If we fail to comply with
these requirements, we would be subject to possible regulatory
action and may be limited in the jurisdictions in which we are
permitted to sell our products and will lose time to market and
potential revenues.
S-16
It is uncertain whether product liability insurance will be
adequate to address product liability claims, or that insurance
against such claims will be affordable or available on acceptable
terms in the future.
Clinical research
involves the testing of new drugs on human volunteers pursuant to a
clinical trial protocol. Such testing involves a risk of liability
for personal injury to or death of patients due to, among other
causes, adverse side effects, improper administration of the new
drug, or improper volunteer behavior. Claims may arise from
patients, clinical trial volunteers, consumers, physicians,
hospitals, companies, institutions, researchers, or others using,
selling, or buying our products, as well as from governmental
bodies. In addition, product liability and related risks are likely
to increase over time, in particular upon the commercialization or
marketing of any products by us or parties with which we enter into
development, marketing, or distribution collaborations. Although we
are contracting for general liability insurance in connection with
our ongoing business, there can be no assurance that the amount and
scope of such insurance coverage will be appropriate and sufficient
in the event any claims arise, that we will be able to secure
additional coverage should we attempt to do so, or that our
insurers would not contest or refuse any attempt by us to collect
on such insurance policies. Furthermore, there can be no assurance
that suitable product liability insurance (at the clinical stage
and/or commercial stage) will continue to be available on terms
acceptable to us or at all, or that, if obtained, the insurance
coverage will be appropriate and sufficient to cover any potential
claims or liabilities.
If the market opportunities
for our current and potential future drug candidates are smaller
than we believe they are, our ability to generate product revenues
will be adversely affected and our business may
suffer.
Our
understanding of the number of people who suffer from SOM resulting
from chemoradiotherapy for the treatment of OPC, whom Validive may
have the potential to treat, is based upon estimates. These
estimates may prove to be incorrect, and new studies may
demonstrate or suggest a lower estimated incidence or prevalence of
this condition. The number of patients in the U.S. or elsewhere may
turn out to be lower than expected, may not be otherwise amenable
to Validive treatment, or treatment-amenable patients may become
increasingly difficult to identify and access, all of which would
adversely affect our business prospects and financial condition. In
particular, the treatable population for Validive may further be
reduced if our estimates of addressable populations are erroneous
or sub-populations of patients within the addressable population do
not derive benefit from Validive.
Risks Related to Our Reliance on Third Parties
Corporate, non-profit, and academic collaborators may take actions
(including lack of effective actions) to delay, prevent, or
undermine the success of our products.
Our
operating and financial strategy for the development, clinical
testing, manufacture, and commercialization of product candidates
is heavily dependent on us entering into collaborations with
corporations, non-profit organizations, academic institutions,
licensors, licensees, and other parties. There can be no assurance
that we will be successful in establishing such collaborations.
Current and future collaborations are and may be terminable at the
sole discretion of the collaborator. The activities of any
collaborator will not be within our direct control and may not be
in our power to influence. There can be no assurance that any
collaborator will perform its obligations to our satisfaction or at
all; that we will derive any revenue, profits, or benefit from such
collaborations; or that any collaborator will not compete with us.
If any collaboration is not pursued, we may require substantially
greater capital to undertake development and commercialization of
our proposed products and may not be able to develop and
commercialize such products effectively, if at all. In addition, a
lack of development and commercialization collaborations may lead
to significant delays in introducing proposed products into certain
markets and/or reduced sales of proposed products in such markets.
Furthermore, current and future collaborators may act deliberately
or inadvertently in ways detrimental to our interests.
The termination of third-party licenses could adversely affect our
rights to important compounds or technologies.
We have
exercised our option to license Validive; as such, Onxeo has the
ability to terminate the license if we breach our obligations under
the license agreement. A termination of the license agreement might
force us to cease developing and/or selling Validive, if it gets to
market. We rely on certain rights to MNPR-101 that we have secured
through a non-exclusive license agreement with XOMA. XOMA, as
licensor, has the ability to terminate the license if we breach our
obligations under the license agreement and do not remedy any such
breach within a set time after receiving written notice of such
breach from XOMA. A termination of the license agreement might
force us to cease developing and/or selling MNPR-101, if it gets to
market.
Data provided by collaborators and other parties upon which we rely
have not been independently verified and could turn out to be
inaccurate, misleading, or incomplete.
We rely
on third-party vendors, scientists, and collaborators to provide us
with significant data and other information related to our
projects, clinical trials, and business. We do not independently
verify or audit all of such data (including possibly material
portions thereof). As a result, such data may be inaccurate,
misleading, or incomplete.
In certain cases, we may need to rely on a single supplier for a
particular manufacturing material or service, and any interruption
in or termination of service by such supplier could delay or
disrupt the commercialization of our products.
We rely
on third-party suppliers for the materials used to manufacture our
compounds. Some of these materials may at times only be available
from one supplier. Any interruption in or termination of service by
such single source suppliers could result in a delay or disruption
in manufacturing until we locate an alternative source of supply.
There can be no assurance that we would be successful in locating
an alternative source of supply or in negotiating acceptable terms
with such prospective supplier.
S-17
Our Validive manufacturer is in the United Kingdom
(“UK”), and it is unknown how they will be impacted by
Brexit; however, if they are negatively impacted, this could
increase our manufacturing costs and adversely impact our financial
condition.
The
UK’s referendum to leave the EU or “Brexit,” has
caused and may continue to cause disruptions to capital and
currency markets worldwide. The full impact of the Brexit decision,
however, remains uncertain. A process of negotiation will determine
the future terms of the UK’s relationship with the EU. During
this period of negotiation and afterwards, our Validive
manufacturer may be negatively affected by interest rate, exchange
rate and other market and economic volatility, as well as
regulatory and political uncertainty. The tax consequences of the
UK’s withdrawal from the EU are uncertain as well. If Brexit
has a detrimental effect on our Validive manufacturer, it could, in
turn, adversely impact our manufacturing costs and financial
condition.
We rely on third parties to conduct our non-clinical studies and
our clinical trials. If these third parties do not successfully
carry out their contractual duties or meet expected deadlines, we
may be unable to obtain regulatory approval for or commercialize
our current product candidates or any future products, on a timely
basis or at all, and our financial condition will be adversely
affected.
We do
not have the ability to independently conduct non-clinical studies
and clinical trials. We rely on medical institutions, clinical
investigators, contract laboratories, collaborative partners and
other third parties, such as contract research organizations or
clinical research organizations, to conduct non-clinical studies
and clinical trials on our product candidates. The third parties
with whom we contract for execution of our non-clinical studies and
clinical trials play a significant role in the conduct of these
studies and trials and the subsequent collection and analysis of
data. However, these third parties are not our employees, and
except for contractual duties and obligations, we have limited
ability to control the amount or timing of resources that they
devote to our programs.
Although we rely on
third parties to conduct our non-clinical studies and clinical
trials, we remain responsible for ensuring that each of our
non-clinical studies and clinical trials is conducted in accordance
with its investigational plan and protocol. Moreover, the FDA, EMA
and other foreign regulatory authorities require us to comply with
regulations and standards, including some regulations commonly
referred to as good clinical practices (“GCPs”), for
conducting, monitoring, recording and reporting the results of
clinical trials to ensure that the data and results are
scientifically credible and accurate, and that the trial subjects
are adequately informed of the potential risks of participating in
clinical trials.
In
addition, the execution of non-clinical studies and clinical
trials, and the subsequent compilation and analyses of the data
produced, requires coordination among various parties. In order for
these functions to be carried out effectively and efficiently, it
is imperative that these parties communicate and coordinate with
one another. Moreover, these third parties may also have
relationships with other commercial entities, some of which may
compete with us. Under certain circumstances, these third parties
may be able to terminate their agreements with us upon short
notice. If the third parties conducting our clinical trials do not
perform their contractual duties or obligations, experience work
stoppages, do not meet expected deadlines, terminate their
agreements with us or need to be replaced, or if the quality or
accuracy of the clinical data they obtain is compromised due to the
failure to adhere to our clinical trial protocols or GCPs, or for
any other reason, we may need to enter into new arrangements with
alternative third parties, which could be difficult, costly or
impossible, and our clinical trials may be extended, delayed or
terminated or may need to be repeated. If any of the foregoing were
to occur, we may not be able to obtain, on a timely basis or at
all, regulatory approval for or to commercialize the product
candidate being tested in such trials, and as a result, our
financial condition will be adversely affected.
Risks Related to Commercialization of Our Product
Candidates
We have no experience as a company in commercializing any product.
If we fail to obtain commercial expertise, upon product approval by
regulatory agencies, our product launch and revenues could be
delayed.
As a
company, we have never obtained regulatory approval for, or
commercialized, any product. Accordingly, we have not yet begun to
build out any sales or marketing capabilities. If we are unable to
establish, or contract for, effective sales and marketing
capabilities, or if we are unable to enter into agreements with
third parties to commercialize our product candidates on favorable
terms or on any reasonable terms at all, we may not be able to
effectively generate product revenues once our product candidates
are approved for marketing. If we fail to obtain commercial
expertise or capabilities, upon drug approval, our product launch
and subsequent revenues could be delayed and /or fail to reach
their commercial potential.
Our product development efforts are at an early stage. We have not
yet undertaken any marketing efforts, and there can be no assurance
that future anticipated market testing and analyses will validate
our marketing strategy. We may need to modify the products, or we
may not be successful in either developing or marketing those
products.
As a
company, we have not completed the development or clinical trials
of any product candidates and, accordingly, have not yet begun to
market or generate revenue from the commercialization of any
products. Obtaining approvals of these product candidates will
require substantial additional research and development as well as
costly clinical trials. There can be no assurance that we will
successfully complete development of our product candidates or
successfully market them. We may encounter problems and delays
relating to research and development, regulatory approval,
intellectual property rights of product candidates, or other
factors. There can be no assurance that our development programs
will be successful, that our product candidates will prove to be
safe and effective in or after clinical trials, that the necessary
regulatory approvals for any product candidates will be obtained,
or, even if obtained, will be as broad as sought or will be
maintained for any period thereafter, that patents will issue on
our patent applications, that any intellectual property protections
we secure will be adequate, or that our collaboration arrangements
will not diminish the value of our intellectual property through
licensing or other arrangements. Furthermore, there can be no
assurance that any product we might market will be received
favorably by customers (whether physicians, payers, patients, or
all three), adequately reimbursed by third-party payers, or that
competitive products will not perform better and/or be marketed
more successfully. Additionally, there can be no assurances that
any future market testing and analyses will validate our marketing
strategies. We may need to seek to modify the product labels
through additional studies in order to be able to market them
successfully to reach their commercial potential.
S-18
If we are unable to establish relationships with licensees or
collaborators to carry out sales, marketing, and distribution
functions or to create effective marketing, sales, and distribution
capabilities, we will be unable to market our products
successfully.
Our
business strategy may include out-licensing product candidates to
or collaborating with larger firms with experience in marketing and
selling pharmaceutical products. There can be no assurance that we
will successfully be able to establish marketing, sales, or
distribution relationships with any third-party, that such
relationships, if established, will be successful, or that we will
be successful in gaining market acceptance for any products we
might develop. To the extent that we enter into any marketing,
sales, or distribution arrangements with third parties, our product
revenues per unit sold are expected to be lower than if we
marketed, sold, and distributed our products directly, and any
revenues we receive will depend upon the efforts of such third
parties.
If we
are unable to establish such third-party marketing and sales
relationships, or choose not to do so, we would have to establish
in-house marketing and sales capabilities. We have no experience in
marketing or selling oncology pharmaceutical products, and
currently have no marketing, sales, or distribution infrastructure
and no experience developing or managing such infrastructure for an
oncology related product. To market any products directly, we would
have to establish a marketing, sales, and distribution force that
has technical expertise and could support a distribution
capability. Competition in the biopharmaceutical industry for
technically proficient marketing, sales, and distribution personnel
is intense and attracting and retaining such personnel may
significantly increase our costs. There can be no assurance that we
will be able to establish internal marketing, sales, or
distribution capabilities or that these capabilities will be
sufficient to meet our needs.
Commercial success of our product candidates will depend on the
acceptance of these products by physicians, payers, and
patients.
Any
product candidate that we may develop may not gain market
acceptance among physicians and patients. Market acceptance of and
demand for any product that we may develop will depend on many
factors, including without limitation:
●
Comparative
superiority of the effectiveness and safety in the treatment of the
disease indication compared to alternative treatments;
●
Less prevalence and
severity of adverse side effects;
●
Potential
advantages over alternative treatments;
●
Convenience and
ease of administration;
●
Sufficient
third-party coverage and/or reimbursement;
●
Strength of sales,
marketing and distribution support; and
●
Our ability to
provide acceptable evidence of safety and efficacy
If any
product candidate developed by us receives regulatory approval but
does not achieve an adequate level of market acceptance by
physicians, payers, and patients, we may generate insufficient,
little, or no product revenue or earn appropriate returns on the
investment of product development costs and may not become
profitable.
Our products may not be accepted for reimbursement or properly
reimbursed by third-party payers.
The
successful commercialization of any products we might develop will
depend substantially on whether the costs of our products and
related treatments are reimbursed at acceptable levels by
government authorities, private healthcare insurers, and other
third-party payers, such as health maintenance organizations.
Reimbursement rates may vary, depending upon the third-party payer,
the type of insurance plan, and other similar or dissimilar
factors. If our products do not achieve adequate reimbursement,
then the number of physician prescriptions of our products may not
be sufficient to make our products profitable.
Comparative
effectiveness research demonstrating benefits of a
competitor’s product could adversely affect the sales of our
product candidates. If third-party payers do not consider our
products to be cost-effective compared to other available
therapies, they may not cover our products as a benefit under their
plans or, if they do, the level of payment may not be sufficient to
allow us to sell our products on a profitable basis.
Adequate
third-party reimbursement may not be available to enable us to
maintain price levels sufficient to realize an appropriate return
on our investment in the product development of that product. In
addition, in the U.S. there is a growing emphasis on comparative
effectiveness research, both by private payers and by government
agencies. To the extent other drugs or therapies are found to be
more effective than our products, payers may elect to cover such
therapies in lieu of our products or reimburse our products at a
lower rate.
The effects of economic and political pressure to lower
pharmaceutical prices are a major threat to the economic viability
of new research-based pharmaceutical products, and any development
along these lines could materially and adversely affect our
prospects.
Emphasis on managed
care and government price controls in the U.S. has increased and we
expect this will continue to increase the pressure on
pharmaceutical pricing. Coverage policies and third-party
reimbursement rates may change at any time. Even if favorable
coverage and reimbursement status is attained for one or more
products for which we receive regulatory approval, less favorable
coverage policies and reimbursement rates may be implemented in the
future.
S-19
Any
development along these lines could materially and adversely affect
our prospects. We are unable to predict what legislative or
regulatory changes relating to the healthcare industry, including
without limitation any changes affecting governmental and/or
private or third-party coverage and reimbursement, may be enacted
in the future, or what effect such legislative or regulatory
changes would have on our business.
If we obtain FDA approval for any of our product candidates, we
will be subject to various federal and state fraud and abuse laws;
these laws may impact, among other things, our proposed sales,
marketing and education programs. Fraud and abuse laws are expected
to increase in breadth and in detail, which will likely increase
our operating costs and the complexity of our programs to insure
compliance with such enhanced laws.
If we
obtain FDA approval for any of our product candidates and begin
commercializing those products in the U.S., our operations may be
directly, or indirectly through our customers, distributors, or
other business partners, subject to various federal and state fraud
and abuse laws, including, without limitation, anti-kickback
statutes and false claims statutes which may increase our operating
costs. These laws may impact, among other things, our proposed
sales, marketing and education programs. In addition, we may be
subject to data privacy and security regulation by both the federal
government and the states in which we conduct
business.
If our operations are found to be in violation of any of the
federal and state fraud and abuse laws or any other governmental
regulations that apply to us, we may be subject to criminal actions
and significant civil monetary penalties, which would adversely
affect our ability to operate our business and our results of
operations.
If our
operations are found to be in violation of any of the federal and
state fraud and abuse laws, including, without limitation,
anti-kickback statutes and false claims statutes or any other
governmental regulations that apply to us, we may be subject to
penalties, including criminal and significant civil monetary
penalties, damages, fines, imprisonment, exclusion from
participation in government healthcare programs, and the
curtailment or restructuring of our operations, any of which could
adversely affect our ability to operate our business and our
results of operations. To the extent that any of our product
candidates are ultimately sold in a foreign country, we may be
subject to similar foreign laws and regulations, which may include,
for instance, applicable post-marketing requirements, including
safety surveillance, anti-fraud and abuse laws, and implementation
of corporate compliance programs and reporting of payments or
transfers of value to healthcare professionals.
Negotiated prices for our products covered by a Part D prescription
drug plan and other government programs will be lower than the
prices we might otherwise obtain.
Government payment
for some of the costs of prescription drugs may increase demand for
our products for which we receive marketing approval; however, any
negotiated prices for our products covered by a Part D prescription
drug plan and other government programs will be lower than the
prices we might otherwise obtain.
Risks Related to Our Intellectual Property
If we and our third-party licensors do not obtain and preserve
protection for our respective intellectual property rights, our
competitors may be able to take advantage of our (and our
licensors’) development efforts to develop competing
drugs.
Our
commercial success will depend in part on obtaining patent
protection for any products and other technologies we might
develop, and successfully defending any patents we obtain against
third-party challenges. We have licensed all intellectual property
related to Validive from Onxeo S.A., a French public company. See
“Business - Partnerships, Licensing and Acquisition”.
The assignment and transfer of the camsirubicin (formerly GPX-150)
patent portfolio from TacticGem, LLC (“TacticGem”) to
us has been completed. We filed and have been granted in the U.S.
and various countries around the world patents for antibodies that
target uPAR for our MNPR-101 program. We have also been granted in
the U.S. and various countries around the world patents to a
specific sequence of amino acids on uPAR, to which our MNPR-101
antibody binds. We are currently prosecuting this patent in other
countries around the world to further protect MNPR-101. The patent
process is subject to numerous risks and uncertainties, and there
can be no assurance that we will be successful in obtaining and
defending patents. See “Business - Intellectual Property
Portfolio and Exclusivity”. These risks and uncertainties
include without limitation the following:
●
Patents that may be
issued or licensed may be challenged, invalidated, or circumvented;
or may not provide any competitive advantage for other
reasons.
●
Our licensors may
terminate or breach our existing or future license agreements,
thereby reducing or preventing our ability to exclude competition;
termination of such license agreements may also subject us to risk
of patent infringement of patents to which we no longer have a
license.
●
Our competitors,
many of which have substantially greater resources than us and have
made significant investments in competing technologies, may seek,
or may already have obtained, patents that will limit, interfere
with, or eliminate our ability to make, use, and sell our potential
products either in the U.S. or in international
markets.
●
As a matter of
public policy regarding worldwide health concerns, there may be
significant pressure on the U.S. government and other international
governmental bodies to limit the scope of domestic and
international patent protection for cancer treatments that prove
successful.
●
Countries other
than the U.S. may have less restrictive patent laws than those
upheld by the U.S. courts; therefore, non-U.S. competitors could
exploit these laws to create, develop, and market competing
products. In some countries, the legal compliance with
pharmaceutical patents, patent applications and other intellectual
property regulations is very weak or actively evaded in some cases
with government aid.
S-20
In
addition, the U.S. Patent and Trademark Office
(“USPTO”) and patent offices in other jurisdictions
have often required that patent applications concerning
pharmaceutical and/or biotechnology-related inventions be limited
or narrowed substantially to cover only the specific innovations
exemplified in the patent application, thereby limiting their scope
of protection against competitive challenges. Thus, even if we or
our licensors are able to obtain patents, the patents may be
substantially narrower than anticipated.
If we
permit our patents to lapse or expire, we will not be protected and
will have less of a competitive advantage. The value of our
products may be greatly reduced if this occurs. Our patents expire
at different times and are subject to the laws of multiple
countries. Some of our patents are currently near expiration and we
may pursue patent term extensions for these where appropriate. See
“Business - Intellectual Property Portfolio and
Exclusivity”.
In
addition to patents, we also rely on trade secrets and proprietary
know-how. While we take measures to protect this information by
entering into confidentiality and invention agreements with our
consultants and collaborators, we cannot provide any assurances
that these agreements will be fully enforceable and will not be
breached, that we will be able to protect ourselves from the
harmful effects of disclosure if they are not fully enforceable or
are breached, that any remedy for a breach will adequately
compensate us, that these agreements will achieve their intended
aims, or that our trade secrets will not otherwise become known or
be independently discovered by competitors. If any of these events
for which we cannot provide assurances occurs, or we otherwise lose
protection for our trade secrets or proprietary know-how, the value
of this information may be greatly reduced.
The patent protection we obtain and preserve for our product
candidates may not be sufficient enough to provide us with any
competitive advantage.
We may
be subject to competition despite the existence of intellectual
property we license or own. We can give no assurances that our
intellectual property claims will be sufficient to prevent third
parties from designing around patents we own or license and
developing and commercializing competitive products. The existence
of competitive products that avoid our intellectual property could
materially adversely affect our operating results and financial
condition. Furthermore, limitations, or perceived limitations, in
our intellectual property may limit the interest of third parties
to partner, collaborate or otherwise transact with us, if third
parties perceive a higher than acceptable risk to commercialization
of our products or future products. When looking at our Validive
patents’ ability to block competition, the protection offered
by our patents may be, to some extent, more limited than the
protection provided by patents claiming the composition of matter
of entirely new chemical structures previously unknown. If a
competitor were able to successfully design around any method of
use and formulation patents we may have now or in the future, our
business and competitive advantage could be adversely
affected.
Patent terms may be inadequate to protect our competitive position
on our product candidates for an adequate amount of
time.
Patents
have a limited lifespan. In the U.S., if all maintenance fees are
timely paid, the natural expiration of a patent is generally 20
years from its earliest U.S. non-provisional filing date. Various
extensions may be available, but the life of a patent, and the
protection it affords, is limited. Even if patents covering our
product candidates are obtained, once the patent life has expired
for a product candidate, we may be open to competition from
competitive medications, including generic medications. Given the
amount of time required for the development, testing and regulatory
review of new product candidates, patents protecting such product
candidates might expire before or shortly after such product
candidates are commercialized. As a result, our owned and licensed
patent portfolio may not provide us with sufficient rights to
exclude others from commercializing product candidates similar or
identical to ours.
Depending upon the
timing, duration and conditions of any FDA marketing approval of
our product candidates, one or more of our U.S. patents may be
eligible for limited patent term extension under the Drug Price
Competition and Patent Term Restoration Act of 1984, referred to as
the Hatch-Waxman Amendments, and similar legislation in the
European Union. The Hatch-Waxman Amendments permit a patent term
extension of up to five years for a patent covering an approved
product as compensation for effective patent term lost during
product development and the FDA regulatory review process. However,
we may not receive an extension if we fail to exercise due
diligence during the testing phase or regulatory review process,
fail to apply within applicable deadlines, fail to apply prior to
expiration of relevant patents or otherwise fail to satisfy
applicable requirements. Moreover, the length of the extension
could be less than we request. Only one patent per approved product
can be extended, the extension cannot extend the total patent term
beyond 14 years from approval and only those claims covering the
approved drug, a method for using it or a method for manufacturing
it may be extended. If we are unable to obtain patent term
extension or the term of any such extension is less than we
request, the period during which we can enforce our patent rights
for the applicable product candidate will be shortened and our
competitors may obtain approval to market competing products
sooner. As a result, our revenue from applicable products could be
reduced. Further, if this occurs, our competitors may take
advantage of our investment in development and trials by
referencing our clinical and preclinical data and launch their
product earlier than might otherwise be the case, and our
competitive position, business, financial condition, results of
operations, and prospects could be materially harmed.
Intellectual property disputes could require us to spend time and
money to address such disputes and could limit our intellectual
property rights.
The
biopharmaceutical industry has been characterized by extensive
litigation regarding patents and other intellectual property
rights, and companies have employed intellectual property
litigation and USPTO post-grant proceedings to gain a competitive
advantage. We may become subject to infringement claims or
litigation arising out of patents and pending applications of our
competitors, or additional interference proceedings declared by the
USPTO to determine the priority and patentability of inventions.
The defense and prosecution of intellectual property suits, USPTO
proceedings, and related legal and administrative proceedings are
costly and time-consuming to pursue, and their outcome is
uncertain. Litigation may be necessary to enforce our issued
patents, to protect our trade secrets and know-how, or to determine
the enforceability, scope, and validity of the proprietary rights
of others. An adverse determination in litigation or USPTO
post-grant and interference proceedings to which we may become a
party could subject us to significant liabilities, require us to
obtain licenses from third parties, or restrict or prevent us from
selling our products in certain markets. Even if a given patent or
intellectual property dispute were settled through licensing or
similar arrangements, our costs associated with such arrangements
may be substantial and could include the payment by us of large
fixed payments and ongoing royalties. Furthermore, the necessary
licenses may not be available on satisfactory terms or at all. Even
where we have meritorious claims or defenses, the costs of
litigation may prevent us from pursuing these claims or defenses
and/or may require extensive financial and personnel resources to
pursue these claims or defenses. In addition, it is possible there
may be defects of form in our current and future patents that could
result in our inability to defend the intended claims. Intellectual
property disputes arising from the aforementioned factors, or other
factors, may materially harm our business.
S-21
We may not be able to enforce our intellectual property rights
throughout the world.
The
laws of some foreign countries do not protect intellectual property
rights to the same extent as the laws of the U.S. Companies have
encountered significant problems in protecting and defending
intellectual property rights in certain foreign jurisdictions. The
legal systems of some countries, particularly developing countries,
do not favor the enforcement of patents and other intellectual
property protection, especially those relating to life sciences.
This could make it difficult for us to stop the infringement of our
patents or the misappropriation of our other intellectual property
rights. For example, many foreign countries have compulsory
licensing laws under which a patent owner must grant licenses to
third parties. In addition, many countries limit the enforceability
of patents against third parties, including government agencies or
government contractors. In these countries, patents may provide
limited or no benefit.
Proceedings to
enforce our patent rights in foreign jurisdictions, whether or not
successful, could result in substantial costs and divert our
efforts and attention from other aspects of our business.
Furthermore, while we intend to protect our intellectual property
rights in our expected significant markets, we cannot ensure that
we will be able to initiate or maintain similar efforts in all
jurisdictions in which we may wish to market Validive or any future
products. Accordingly, our efforts to protect our intellectual
property rights in such countries may be inadequate. In addition,
changes in the law and legal decisions by courts in the U.S. and
foreign countries may affect our ability to obtain and enforce
adequate intellectual property protection for our products and
technology.
If we are unable to protect the confidentiality of our trade
secrets, our business and competitive position would be
harmed.
In
addition to seeking patent protection, we also rely on trade
secrets, including unpatented know-how, technology and other
proprietary information, to maintain our competitive position. We
seek to protect these trade secrets, in part, by entering into
non-disclosure and confidentiality agreements with parties who have
access to them. Despite these efforts, these parties may breach the
agreements and disclose our proprietary information, including our
trade secrets, and we may not be able to obtain adequate remedies
for such breaches. Enforcing a claim that a party illegally
disclosed or misappropriated a trade secret is difficult, expensive
and time-consuming, and the outcome is unpredictable. In addition,
some courts inside and outside the U.S., including in foreign
jurisdictions, are less willing or unwilling to protect trade
secrets. If any of our trade secrets were to be lawfully obtained
or independently developed by a competitor, we would have no right
to prevent them from using that technology or information to
compete with us. If any of our trade secrets were to be disclosed
to or independently developed by a competitor, our competitive
position would be harmed.
Changes to the patent law in the U.S. and other jurisdictions could
diminish the value of patents in general, thereby impairing our
ability to protect our product candidates.
As is
the case with other biopharmaceutical companies, our success is
heavily dependent on intellectual property, particularly patents.
Obtaining and enforcing patents in the biopharmaceutical industry
involves both technological and legal complexity. Therefore,
obtaining and enforcing biopharmaceutical patents is costly, time
consuming and inherently uncertain. In addition, the U.S. has
recently enacted and is currently implementing wide ranging patent
reform legislation. The U.S. Supreme Court has ruled on several
patent cases in recent years, either narrowing the scope of patent
protection available in certain circumstances or weakening the
rights of patent owners in certain situations. In addition to
increasing uncertainty with regard to our ability to obtain patents
in the future, this combination of events has created uncertainty
with respect to the value of patents once obtained. Depending on
future actions by the U.S. Congress, the federal courts and the
USPTO, as well as other jurisdictions around the world, the laws
and regulations governing patents could change in unpredictable
ways that would weaken our ability to obtain new patents or to
enforce our existing patents and patents that we might obtain in
the future.
Obtaining and maintaining our patent protection depends on
compliance with various procedural, document submission, fee
payment and other requirements imposed by governmental patent
agencies, and our patent protection could be reduced or eliminated
for non-compliance with these requirements.
The
USPTO and various foreign governmental patent agencies require
compliance with a number of procedural, documentary, fee payment
and other provisions during the patent process. There are
situations in which noncompliance can result in abandonment or
lapse of a patent or patent application, resulting in partial or
complete loss of patent rights in the relevant jurisdiction. In
such an event, competitors might be able to enter the market
earlier than would otherwise have been the case.
If we fail to comply with our obligations under any license,
collaboration or other intellectual property-related agreements, we
may be required to pay damages and could lose intellectual property
rights that may be necessary for developing, commercializing and
protecting our current or future technologies or drug candidates or
we could lose certain rights to grant sublicenses.
Any
license, collaboration or other intellectual property-related
agreements impose, and any future license, collaboration or other
intellectual property-related agreements we enter into are likely
to impose, various development, commercialization, funding,
milestone, royalty, diligence, sublicensing, insurance, patent
prosecution and enforcement or other obligations on us. If we
breach any of these obligations, or use the intellectual property
licensed to us in an unauthorized manner, we may be required to pay
damages and the licensor may have the right to terminate the
license. In spite of our best efforts, any of our future licensors
might conclude that we have materially breached our license
agreements and might therefore terminate the license agreements,
thereby removing our ability to develop and commercialize products
and technologies covered by these license agreements. Any license
agreements we enter into may be complex, and certain provisions in
such agreements may be susceptible to multiple interpretations. The
resolution of any contract interpretation disagreement that may
arise could narrow what we believe to be the scope our rights to
the relevant intellectual property or technology, or increase what
we believe to be our financial or other obligations under the
relevant agreement, either of which could have a material adverse
effect on our business, financial condition, results of operations,
and prospects.
S-22
We may
seek to obtain licenses from licensors in the future, however, we
may be unable to obtain any such licenses at a reasonable cost or
on reasonable terms, if at all. In addition, if any of our future
licensors terminate any such license agreements, such license
termination could result in our inability to develop, manufacture
and sell products that are covered by the licensed technology or
could enable a competitor to gain access to the licensed
technology. Any of these events could have a material adverse
effect on our competitive position, business, financial condition,
results of operations, and ability to achieve
profitability.
Furthermore, we may
not have the right to control the preparation, filing, prosecution,
maintenance, enforcement and defense of patents and patent
applications that we license from third parties. Therefore, we
cannot be certain that these patents and patent applications will
be prepared, filed, prosecuted, maintained, enforced and defended
in a manner consistent with the best interests of our business. If
our future licensors fail to prosecute, maintain, enforce and
defend patents we may in-license, or lose rights to licensed
patents or patent applications, our license rights may be reduced
or eliminated. In such circumstances, our right to develop and
commercialize any of our products or drug candidates that is the
subject of such licensed rights could be materially adversely
affected. In certain circumstances, our licensed patent rights are
subject to our reimbursing our licensors for their patent
prosecution and maintenance costs.
Moreover, our
licensors may own or control intellectual property that has not
been licensed to us and, as a result, we may be subject to claims,
regardless of their merit, that we are infringing, misappropriating
or otherwise violating the licensor’s intellectual property
rights and the amount of any damages or future royalty obligations
that would result, if any such claims were successful, would depend
on the technology and intellectual property we use in products that
we successfully develop and commercialize, if any. Therefore, even
if we successfully develop and commercialize products, due to such
obligations, we may be unable to achieve or maintain
profitability.
Third parties
may initiate legal proceedings alleging that we are infringing,
misappropriating or otherwise violating their intellectual property
rights, the outcome of which would be uncertain and could have a
material adverse impact on the success of our
business.
Our
commercial success depends, in part, upon our ability or the
ability of any of our future collaborators to develop, manufacture,
market and sell our current or any future drug candidates and to
use our proprietary technologies without infringing,
misappropriating or otherwise violating the proprietary and
intellectual property rights of third parties. The biotechnology
and pharmaceutical industries are characterized by extensive and
complex litigation regarding patents and other intellectual
property rights.
We or
any of our future licensors or strategic partners, may be party to,
or be threatened with, adversarial proceedings or litigation
regarding intellectual property rights with respect to our current
or any potential future drug candidates and technologies, including
derivation, reexamination, inter partes review, post-grant review
or interference proceedings before the USPTO and similar
proceedings in jurisdictions outside of the U.S. such as opposition
proceedings. If we or our licensors or strategic partners are
unsuccessful in any interference proceedings or other priority or
validity disputes (including through any patent oppositions) to
which we or they are subject, we may lose valuable intellectual
property rights through the loss of one or more patents or our
patent claims may be narrowed, invalidated, or held unenforceable.
In some instances, we may be required to indemnify our licensors or
strategic partners for the costs associated with any such
adversarial proceedings or litigation. Third parties may also
assert infringement, misappropriation or other claims against us,
our licensors or our strategic partners based on existing patents
or patents that may be granted in the future, as well as other
intellectual property rights, regardless of their merit. There is a
risk that third parties may choose to engage in litigation or other
adversarial proceedings with us, our licensors or our strategic
partners to enforce or otherwise assert their patent rights or
other intellectual property rights. Even if we believe such claims
are without merit, a court of competent jurisdiction could hold
that these third-party patents and other intellectual property
rights are valid, enforceable and infringed, which could have a
material adverse impact on our ability to utilize our developed
technologies or to commercialize our current or any future drug
candidates deemed to be infringing. In order to successfully
challenge the validity of any such U.S. patent in federal court, we
would need to overcome a presumption of validity by presenting
clear and convincing evidence of invalidity. There is no assurance
that a court of competent jurisdiction, even if presented with
evidence we believe to be clear and convincing, would invalidate
the claims of any such U.S. patent.
Further, we cannot
guarantee that we will be able to successfully settle or otherwise
resolve such adversarial proceedings or litigation. If we are
unable to successfully settle future claims on terms acceptable to
us, we may be required to engage in or to continue costly,
unpredictable and time-consuming litigation and may be prevented
from or experience substantial delays in marketing our drug
candidates. If we or any of our licensors or strategic partners are
found to infringe, misappropriate or violate a third-party patent
or other intellectual property rights, we could be required to pay
damages, including treble damages and attorney’s fees, if we
are found to have willfully infringed. In addition, we, or any of
our licensors or strategic partners may choose to seek, or be
required to seek, a license from a third-party, which may not be
available on commercially reasonable terms, if at all. Even if a
license can be obtained on commercially reasonable terms, the
rights may be non-exclusive, which could give our competitors
access to the same technology or intellectual property rights
licensed to us, and we could be required to make substantial
licensing and royalty payments. We also could be forced, including
by court order, to cease utilizing, developing, manufacturing and
commercializing our developed technologies or drug candidates
deemed to be infringing. We may be forced to redesign current or
future technologies or products. Any of the foregoing could have a
material adverse effect on our ability to generate revenue or
achieve profitability and possibly prevent us from generating
revenue sufficient to sustain our operations.
In
addition, we or our licensors or strategic partners may find it
necessary to pursue claims or to initiate lawsuits to protect or
enforce our patent or other intellectual property rights. If we or
our licensors or strategic partners were to initiate legal
proceedings against a third-party to enforce a patent covering one
of our drug candidates or our developed technology, the defendant
could counterclaim that such patent is invalid or unenforceable. In
patent litigation in the U.S., defendant counterclaims alleging
invalidity or unenforceability are commonplace. Grounds for a
validity challenge could be an alleged failure to meet any of
several statutory requirements, for example, claiming patent-
ineligible subject matter, lack of novelty, indefiniteness, lack of
written description, non-enablement, anticipation or obviousness.
Grounds for an unenforceability assertion could be an allegation
that someone connected with prosecution of the patent withheld
relevant information from the USPTO or made a misleading statement
during prosecution. The outcome of such invalidity and
unenforceability claims is unpredictable. With respect to the
validity question, for example, we cannot be certain that there is
no invalidating prior art of which we or our licensors or strategic
partners and the patent examiner were unaware during prosecution.
If a defendant were to prevail on a legal assertion of invalidity
or unenforceability, we could lose at least part, and perhaps all,
of the patent protection for one or more of our drug candidates.
The narrowing or loss of our owned and licensed patent claims could
limit our ability to stop others from using or commercializing
similar or identical technologies and products. All of these events
could have a material adverse effect on our business, financial
condition, results of operations and prospects. Patent and other
intellectual property rights also will not protect our drug
candidates and technologies if competitors or third parties design
around such drug candidates and technologies without legally
infringing, misappropriating or violating our patent or other
intellectual property rights.
S-23
The
cost to us in defending or initiating any litigation or other
proceedings relating to our patent or other intellectual property
rights, even if resolved in our favor, could be substantial, and
any litigation or other proceedings would divert our
management’s attention and distract our personnel from their
normal responsibilities. Such litigation or proceedings could
materially increase our operating losses and reduce the resources
available for development activities or any future sales, marketing
or distribution activities. We may not have sufficient financial or
other resources to conduct such litigation or proceedings
adequately. Some of our competitors may be able to more effectively
sustain the costs of complex patent litigation because they have
substantially greater resources. Uncertainties resulting from the
initiation and continuation of patent litigation or other
proceedings could delay our research and development efforts and
materially limit our ability to continue our operations.
Furthermore, because of the substantial amount of discovery
required in connection with certain such proceedings, there is a
risk that some of our confidential information could be compromised
by disclosure. In addition, there could be public announcements of
the results of hearings, motions or other interim proceedings or
developments and if securities analysts or investors perceive these
results to be negative, such announcements could have a material
adverse effect on the price of our common stock.
Intellectual property rights of third parties could adversely
affect our ability to commercialize our current or future
technologies or drug candidates, and we might be required to
litigate or obtain licenses from third parties to develop or market
our current or future technologies or drug candidates, which may
not be available on commercially reasonable terms, or at
all.
There
are numerous companies that have pending patent applications and
issued patents broadly covering immune-therapies generally or
covering small molecules directed against the same targets as, or
targets similar to, those we are pursuing. Our competitive position
may materially suffer if patents issued to third parties or other
third- party intellectual property rights cover our current or
future technologies, drug candidates or elements thereof, or our
manufacture or uses relevant to our development plans. In such
cases, we may not be in a position to develop or commercialize
current or future technologies or drug candidates unless we
successfully pursue litigation to nullify or invalidate the
third-party intellectual property rights concerned, or enter into a
license agreement with the intellectual property rights holder, if
available on commercially reasonable terms. There may be issued
patents of which we are not aware, held by third parties that, if
found to be valid and enforceable, could be alleged to be infringed
by our current or future technologies or drug candidates. There
also may be pending patent applications of which we are not aware
that may result in issued patents, which could be alleged to be
infringed by our current or future technologies or drug candidates.
Should such an infringement claim be successfully brought, we may
be required to pay substantial damages or be forced to abandon our
current or future technologies or drug candidates or to seek a
license from any patent holders. No assurances can be given that a
license will be available on commercially reasonable terms, if at
all.
Third-party
intellectual property rights holders may also actively bring
infringement, misappropriation or other claims alleging violations
of intellectual property rights against us. We cannot guarantee
that we will be able to successfully settle or otherwise resolve
such claims. If we are unable to successfully settle future claims
on terms acceptable to us, we may be required to engage in or to
continue costly, unpredictable and time-consuming litigation and
may be prevented from, or experience substantial delays in,
marketing our drug candidates. If we fail in any such dispute, in
addition to being forced to pay damages, we may be temporarily or
permanently prohibited from commercializing any of our current or
future technologies or drug candidates that are held to be
infringing, misappropriating or otherwise violating third-party
intellectual property rights. We might, if possible, also be forced
to redesign current or future technologies or drug candidates so
that we no longer infringe, misappropriate or violate the
third-party intellectual property rights. Any of these events, even
if we were ultimately to prevail, could require us to divert
substantial financial and management resources that we would
otherwise be able to devote to our business, which could have a
material adverse effect on our financial condition and results of
operations.
Risks Related to Our Business Operations and
Industry
As a recently established
entity, we have a limited operating history.
As of
January 13, 2020, we have engaged exclusively in acquiring
pharmaceutical product candidates, licensing rights to product
candidates and entering into collaboration agreements with respect
to key services or technologies for our drug product development,
and have not completed any clinical trials, received any
governmental approvals, brought any product to market, manufactured
products in clinical or commercial quantities or sold any
pharmaceutical products. As a company we have limited experience in
negotiating, establishing, and maintaining strategic relationships,
conducting clinical trials, and managing the regulatory approval
process, all of which will be necessary if we are to be successful.
Our lack of experience in these critical areas makes it difficult
for a prospective investor to evaluate our abilities and increases
the risk that we will fail to successfully execute our
strategies.
Furthermore, if our
business grows rapidly, our operational, managerial, legal, and
financial resources will be strained. Our development will require
continued improvement and expansion of our management team and our
operational, managerial, legal, and financial systems and
controls.
In the
normal course of business, we have evaluated and expect to evaluate
potential acquisitions and/or licenses of patents, compounds, and
technologies that our management believes could complement or
expand our business. We have limited history of conducting
acquisitions and negotiating and acquiring licenses. In the event
that we identify an acquisition or license candidate we find
attractive, there is no assurance that we will be successful in
negotiating an agreement to acquire or license, or in financing or
profitably exploiting, such patents, compounds, or technologies.
Furthermore, such an acquisition or license could divert management
time and resources away from other activities that would further
our current business development.
S-24
If we lose key management leadership, and/or scientific personnel,
and if we cannot recruit qualified employees, managers, directors,
officers, or other significant personnel, we may experience program
delays and increases in compensation costs, and our business may be
materially disrupted.
Our
future success is highly dependent on the continued service of
principal members of our management, leadership, and scientific
personnel, who are able to terminate their employment with us at
any time and may be able to compete with us. The loss of any of our
key management, leadership, or scientific personnel including, in
particular, Christopher M. Starr, our Executive Chairman of the
Board of Directors (referred to as the “Board”),
Chandler D. Robinson, our President and CEO, and Andrew P. Mazar,
our Executive Vice President of Research and Development and Chief
Scientific Officer, could materially disrupt our business and
materially delay or prevent the successful product development and
commercialization of our product candidates. We have employment
agreements with Dr. Robinson and Dr. Mazar which have no term but
are for at-will employment, meaning the executives have the ability
to terminate their employment at any time. We do not have an
employment agreement with Dr. Starr.
Our
future success will also depend on our continuing ability to
identify, hire, and retain highly skilled personnel for all areas
of the organization. Competition in the biopharmaceutical industry
for scientifically and technically qualified personnel is intense,
and we may be unsuccessful in identifying, hiring, and retaining
qualified personnel. Our continued requirement to identify, hire,
and retain highly competent personnel may cause our compensation
costs to increase materially.
We will incur increased costs as a result of operating as a stock
trading public company, and our management will be required to
devote substantial time to new compliance initiatives and corporate
governance practices.
As a
stock trading public company, and particularly after we are no
longer an emerging growth company, we will incur significant legal,
accounting and other expenses that we did not incur as a private
company. The Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform
and Consumer Protection Act, the listing requirements of the Nasdaq
Capital Market and other applicable securities rules and
regulations impose various requirements on stock trading public
companies, including establishment and maintenance of effective
disclosure and financial controls and corporate governance
practices. Our management and other personnel will need to devote a
substantial amount of time to these compliance initiatives.
Moreover, these rules and regulations will increase our legal and
financial compliance costs and will make some activities more
time-consuming and costly. For example, we expect that these rules
and regulations may make it more difficult and more expensive for
us to obtain director and officer liability insurance, which in
turn could make it more difficult for us to attract and retain
qualified members of our board of directors. However, these rules
and regulations are often subject to varying interpretations, in
many cases due to their lack of specificity, and, as a result,
their application in practice may evolve over time as new guidance
is provided by regulatory and governing bodies. This could result
in continuing uncertainty regarding compliance matters and higher
costs necessitated by ongoing revisions to disclosure and
governance practices.
Despite ongoing compliance training and periodic education, our
employees and consultants may engage in misconduct or other
improper activities, including noncompliance with regulatory
standards and requirements, which could result in delays or
terminations of our development programs and adversely affect our
business.
Although we
regularly train our employees on compliance and we are aware of no
misconduct or improper activities to date, we are exposed to the
risk of employee or consultant fraud or other misconduct.
Misconduct by our employees or consultants could include
intentional failures to: comply with FDA regulations; provide
accurate information to the FDA; comply with manufacturing
standards; comply with federal and state healthcare fraud and abuse
laws and regulations; report financial information or data
accurately or disclose unauthorized activities to us. In
particular, sales, marketing and business arrangements in the
healthcare industry are subject to extensive laws and regulations
intended to prevent fraud, kickbacks, self-dealing and other
abusive practices. These laws and regulations may restrict or
prohibit a wide range of pricing, discounting, marketing and
promotion, sales commission, customer incentive programs and other
business arrangements. Employee and consultant misconduct could
also involve the improper use of information obtained in the course
of clinical trials, which could result in regulatory sanctions and
serious harm to our reputation. It is not always possible to
identify and deter such misconduct, and the precautions we take to
detect and prevent this activity may not be effective in
controlling unknown or unmanaged risks or losses or in protecting
us from governmental investigations or other actions or lawsuits
stemming from a failure to be in compliance with such laws or
regulations. If any such actions are instituted against us, and we
are not successful in defending ourselves or asserting our rights,
those actions could have a significant impact on our business,
including the imposition of significant fines or other sanctions.
Such actions could adversely affect our business including delaying
or terminating one or more of our development
programs.
S-25
We are an emerging growth company and we cannot be certain if the
reduced disclosure requirements applicable to emerging growth
companies will make our Common Stock less attractive to
investors.
We are
an emerging growth company. Under the Jumpstart Our Business
Startups Act of 2012 (the “JOBS Act”), emerging growth
companies can delay adopting new or revised accounting standards
until such time as those standards apply to private companies. We
have irrevocably elected to opt out of this provision and, as a
result, we will comply with new or revised accounting standards
when they are required to be adopted by public companies that are
not emerging growth companies.
For as
long as we continue to be an emerging growth company, we also
intend to take advantage of certain other exemptions from various
reporting requirements that are applicable to other public
companies including, but not limited to, reduced disclosure
obligations regarding executive compensation in our periodic
reports and proxy statements, exemptions from the requirements of
holding a nonbinding advisory stockholder vote on executive
compensation and any golden parachute payments not previously
approved, exemption from the requirement of auditor attestation in
the assessment of our internal control over financial reporting and
exemption from any requirement that may be adopted by the Public
Company Accounting Oversight Board regarding mandatory audit firm
rotation or a supplement to the auditor’s report providing
additional information about the audit and the financial statements
(auditor discussion and analysis). If we do take advantage of these
exemptions, the information that we provide stockholders will be
different than what is available with respect to other public
companies. We cannot predict if investors will find our common
stock less attractive because we will rely on these exemptions. If
investors find our common stock less attractive as a result of our
status as an emerging growth company, if and when our stock becomes
publicly traded, there may be less liquidity for our common stock
and our stock price may be more volatile.
We will
remain an emerging growth company until the earliest of (1) the
last day of the year (a) following the fifth anniversary of the
completion of our initial public offering, (b) in which we have
total annual gross revenue of at least $1.07 billion or (c) in
which we are deemed to be a large accelerated filer, which means
the market value of our common stock that is held by non-affiliates
exceeds $700 million as of the prior June 30th, and (2) the date on
which we have issued more than $1.0 billion in non-convertible debt
securities during the prior three-year period.
Competition and technological change may make our product
candidates less competitive or obsolete.
The
biopharmaceutical industry is subject to rapid technological
change. We have many potential competitors, including major drug
and chemical companies, specialized biopharmaceutical firms,
universities and other research institutions. These companies,
firms, and other institutions may develop products that are more
effective than our product candidates or that would make our
product candidates less competitive or obsolete. Many of these
companies, firms, and other institutions have greater financial
resources than us and may be better able to withstand and respond
to adverse market conditions within the biopharmaceutical industry,
including without limitation the lengthy regulatory approval
process for product candidates.
We face significant competition from other biotechnology and
pharmaceutical companies, and our operating results will suffer if
we fail to compete effectively.
The
biotechnology and pharmaceutical industries are characterized by
rapidly advancing technologies, intense competition and a strong
emphasis on proprietary products. While we believe we have
significant competitive advantages with our expertise in small
molecules and biologics, and rare disease clinical development,
along with a strong intellectual property portfolio, we currently
face and will continue to face competition for our drug development
programs from companies that target SOM, are developing doxorubicin
analogs/replacement, and are targeting uPAR. The competition is
likely to come from multiple sources, including larger
pharmaceutical companies, biotechnology companies and academia.
Accordingly, our competitors may have more resources and be more
successful than us in obtaining approval for treatments and
achieving widespread market acceptance. For any products that we
may ultimately commercialize, not only will we compete with any
existing therapies and those therapies currently in development, we
will have to compete with new therapies that may become available
in the future.
We
may engage in strategic transactions that could impact our
liquidity, increase our expenses and present significant
distractions to our management.
From
time to time, we may consider strategic transactions, such as
acquisitions of companies, asset purchases, and out-licensing or
in-licensing of products, product candidates or technologies.
Additional potential transactions that we may consider include a
variety of different business arrangements, including spin-offs,
strategic partnerships, joint ventures, restructurings,
divestitures, business combinations and investments. Any such
transaction will require us to incur non-recurring or other
charges, may increase our near- and long-term expenditures and may
pose significant integration challenges or disrupt our management
or business, which could adversely affect our operations and
financial results. For example, these transactions may entail
numerous operational and financial risks, including:
●
exposure to unknown
liabilities;
●
disruption of our
business and diversion of our management's time and attention in
order to develop acquired products, product candidates or
technologies;
●
incurrence of
substantial debt or dilutive issuances of equity securities to pay
for acquisitions;
●
higher-than-expected
acquisition and integration costs;
●
write-downs of
assets, goodwill or impairment charges;
●
increased
amortization expenses;
●
difficulty and cost
in combining the operations and personnel of any acquired
businesses with our operations and personnel;
●
impairment of
relationships with key suppliers or customers of any acquired
businesses due to changes in management and ownership;
and
●
inability to retain
key employees of any acquired businesses.
S-26
Accordingly,
although there can be no assurance that we will undertake or
successfully complete any transactions of the nature described
above, any transactions that we do complete may be subject to the
foregoing or other risks, and could have a material adverse effect
on our business, results of operations, financial condition and
prospects.
If product liability lawsuits are brought against us, we may incur
substantial costs to defend them and address any damages awarded,
and demand for our products could be reduced as a result of such
lawsuits.
The
testing and marketing of medical products is subject to an inherent
risk of product liability claims, including a possibility in some
states for product liability claims being made based on generic
copies of our drugs. Since we currently are not sponsoring any
clinical trials, we do not have product liability insurance
coverage, but plan to obtain appropriate coverage when we enroll
patients in a Validive or other clinical trial, assuming the
coverage is available at a commercially reasonable cost, if
available at all. Regardless of their merit or eventual outcome,
product liability claims may result in:
●
withdrawal of
clinical trial volunteers;
●
decreased demand
for our products when approved;
●
injury to our
reputation and significant, adverse media attention;
and
●
potentially
significant litigation costs, including without limitation, any
damages awarded to the plaintiffs if we lose or settle
claims.
Our business and operations are
vulnerable to computer system failures, cyber-attacks or
deficiencies in our cyber-security, which could increase our
expenses, divert the attention of our management and key personnel
away from our business operations and adversely affect our results
of operations.
Despite
the implementation of security measures, our internal computer
systems, and those of third parties on which we rely, are to damage
from: computer viruses; malware; natural disasters; terrorism; war;
telecommunication and electrical failures; cyber-attacks or
cyber-intrusions over the Internet; attachments to emails; persons
inside our organization; or persons with access to systems inside
our organization. The risk of a security breach or disruption,
particularly through cyber-attacks or cyber intrusion, including by
computer hackers, foreign governments, and cyber terrorists, has
generally increased as the number, intensity and sophistication of
attempted attacks and intrusions from around the world have
increased. If such an event were to occur and cause interruptions
in our operations, it could result in a material disruption of our
product development programs. For example, the loss of clinical
trial data from completed or ongoing or planned clinical trials
could result in delays in our regulatory approval efforts and
significantly increase our costs to recover or reproduce the data.
To the extent that any disruption or security breach was to result
in a loss of or damage to our data or applications, or
inappropriate disclosure of confidential or proprietary
information, we could incur material legal claims and liability,
and damage to our reputation, and the further development of our
product candidates could be delayed. We could be forced to expend
significant resources in response to a cyber security breach,
including repairing system damage, increasing cyber security
protection costs by deploying additional personnel and protection
technologies, paying regulatory fines and resolving legal claims
and regulatory actions, all of which would increase our expenses,
divert the attention of our management and key personnel away from
our business operations and adversely affect our results of
operations.
Failure to comply with health and data protection laws and
regulations could lead to government enforcement actions (which
could include civil or criminal penalties), private litigation or
adverse publicity and could negatively affect our operating results
and business.
We and
our current and any of our future collaborators may be subject to
federal, state and foreign data protection laws and regulations
(i.e., laws and regulations that address privacy and data
security). In the U.S., numerous federal and state laws and
regulations, including federal health information privacy laws
(e.g., the Health Insurance Portability and Accountability Act
(“HIPAA”), as amended by the Health Information
Technology for Economic and Clinical Health Act
(“HITECH”)), state data breach notification laws, state
health information privacy laws and federal and state consumer
protection laws (e.g., Section 5 of the Federal Trade Commission
Act), that govern the collection, use, disclosure and protection of
health-related and other personal information could apply to our
operations or the operations of our collaborators. In addition, we
may obtain health information from third parties (including
research institutions from which we obtain clinical trial data)
that are subject to privacy and security requirements under HIPAA,
as amended by HITECH, or other privacy and data security laws.
Depending on the facts and circumstances, we could be subject to
criminal penalties if we knowingly obtain, use, or disclose
individually identifiable health information maintained by a
HIPAA-covered entity in a manner that is not authorized or
permitted by HIPAA.
International data
protection laws, including Regulation 2016/679, known as the
General Data Protection Regulation (“GDPR”) may also
apply to health-related and other personal information obtained
outside of the U.S. The GDPR went into effect on May 25, 2018. The
GDPR introduced new data protection requirements in the EU, as well
as potential fines for non-compliant companies of up to the greater
of €20 million or 4% of annual global revenue. The regulation
imposes numerous new requirements for the collection, use, storage
and disclosure of personal information, including more stringent
requirements relating to consent and the information that must be
shared with data subjects about how their personal information is
used, the obligation to notify regulators and affected individuals
of personal data breaches, extensive new internal privacy
governance obligations and obligations to honor expanded rights of
individuals in relation to their personal information (e.g., the
right to access, correct and delete their data). In addition, the
GDPR includes restrictions on cross-border data transfers. The GDPR
increased our responsibility and liability in relation to personal
data that we process where such processing is subject to the GDPR,
and we may be required to put in place additional mechanisms to
ensure compliance with the GDPR, including as implemented by
individual countries. Further, the United Kingdom’s vote in
favor of exiting the EU, often referred to as Brexit, has created
uncertainty with regard to data protection regulation in the United
Kingdom. In particular, it is unclear how data transfers to and
from the United Kingdom will be regulated.
S-27
In
addition, California recently enacted the California Consumer
Privacy Act (“CCPA”), which creates new individual
privacy rights for California consumers (as defined in the law) and
places increased privacy and security obligations on entities
handling personal data of consumers or households. The CCPA will
require covered companies to provide new disclosure to consumers
about such companies’ data collection, use and sharing
practices, provide such consumers new ways to opt-out of certain
sales or transfers of personal information, and provide consumers
with additional causes of action. The CCPA goes into effect on
January 1, 2020, and the California Attorney General may bring
enforcement actions for violations beginning July 1, 2020. The CCPA
was amended on September 23, 2018, and it remains unclear what, if
any, further modifications will be made to this legislation or how
it will be interpreted. As currently written, the CCPA may impact
our business activities and exemplifies the vulnerability of our
business to the evolving regulatory environment related to personal
data and protected health information.
Compliance with
U.S. and international data protection laws and regulations could
require us to take on more onerous obligations in our contracts,
restrict our ability to collect, use and disclose data, or in some
cases, impact our ability to operate in certain jurisdictions.
Failure to comply with U.S. and international data protection laws
and regulations could result in government enforcement actions
(which could include civil or criminal penalties), private
litigation or adverse publicity and could negatively affect our
operating results and business.
If we, our CROs or our IT vendors experience security or data
privacy breaches or other unauthorized or improper access to, use
of, or destruction of personal data, we may face costs, significant
liabilities, harm to our brand and business
disruption.
In
connection with our drug research and development efforts, we or
our CROs may collect and use a variety of personal data, such as
names, mailing addresses, email addresses, phone numbers and
clinical trial information. Although we have extensive measures in
place to prevent the sharing and loss of patient data in our
clinical trial processes associated with our developed technologies
and drug candidates, any failure to prevent or mitigate security
breaches or improper access to, use of, or disclosure of our
clinical data or patients’ personal data could result in
significant liability under state (e.g., state breach notification
laws), federal (e.g., HIPAA, as amended by HITECH), and
international laws (e.g., the GDPR). Any failure to prevent or
mitigate security breaches or improper access to, use of, or
disclosure of our clinical data or patients’ personal data
may cause a material adverse impact to our reputation, affect our
ability to conduct new studies and potentially disrupt our
business. We may also rely on third-party IT vendors to host or
otherwise process some of our data and that of users, and any
failure by such IT vendor to prevent or mitigate security breaches
or improper access to or disclosure of such information could have
similarly adverse consequences for us. If we are unable to prevent
or mitigate the impact of such security or data privacy breaches,
we could be exposed to litigation and governmental investigations,
which could lead to a potential disruption to our
business.
If we do not comply with laws regulating the protection of the
environment and health and human safety, our business could be
adversely affected.
Our
research and development and drug candidates and future commercial
manufacturing may involve the use of hazardous materials and
various chemicals. We currently do not maintain a research
laboratory, but we engage third-party research organizations and
manufacturers to conduct our preclinical studies, clinical trials
and manufacturing. These third-party laboratories and manufacturers
are subject to federal, state and local laws and regulations
governing the use, manufacture, storage, handling and disposal of
these hazardous materials. We must rely on the third parties’
procedures for storing, handling and disposing of these materials
in their facilities to comply with the relevant guidelines of the
states in which they operate and the Occupational Safety and Health
Administration of the U.S. Department of Labor. Although we believe
that their safety procedures for handling and disposing of these
materials comply with the standards mandated by applicable
regulations, the risk of accidental contamination or injury from
these materials cannot be eliminated. If an accident occurs, this
could result in significant delays in our development. We are also
subject to numerous environmental, health and workplace safety laws
and regulations. Although we maintain workers’ compensation
insurance to cover us for costs and expenses we may incur due to
injuries to our employees, this insurance may not provide adequate
coverage against potential liabilities. Additional federal, state
and local laws and regulations affecting our operations may be
adopted in the future. We may incur substantial costs to comply
with, and substantial fines or penalties if we violate, any of
these laws or regulations.
We have limited
the liability of and indemnified our directors and
officers.
Although our
directors and officers are accountable to us and must exercise good
faith, good business judgement, and integrity in handling our
affairs, our Second Amended and Restated Certificate of
Incorporation (the “Certificate of Incorporation”),
provides that our directors will be indemnified to the fullest
extent permitted under Delaware law. As a result, our stockholders
may have fewer rights against our directors than they would have
absent such provisions in our Certificate of Incorporation, and a
stockholder’s ability to seek and recover damages for a
breach of fiduciary duties may be reduced or restricted. Delaware
law allows indemnification of members of our Board (each a
“Member”), if such Board Member (a) has acted in good
faith, in a manner the Board Member reasonably believes to be in or
not opposed to our best interests, and (b) with respect to any
criminal action or proceeding, if the Board Member had no
reasonable cause to believe the conduct was unlawful.
Pursuant to the
Certificate of Incorporation, each director and (to the extent
approved by our Board) each of our officers who is made a party to
a legal proceeding because he or she is or was a Board Member or
officer, is indemnified by us from and against any and all
liability, except that we may not indemnify a Board Member or
officer: (a) for any liability incurred in a proceeding in which
such person is adjudged liable to Monopar or is subjected to
injunctive relief in favor of Monopar; (b) for acts or omissions
that involve intentional misconduct or a knowing violation of law,
fraud or gross negligence; (c) for unlawful distributions; (d) for
any transaction for which such Board Member or officer received a
personal benefit or as otherwise prohibited by or as may be
disallowed under Delaware law; or (e) with respect to any dispute
or proceeding between us and such Board Member or officer unless
such indemnification has been approved by a disinterested majority
of Board Members or by a majority in interest of disinterested
stockholders. We are required to pay or reimburse attorney’s
fees and expenses of a Board Member seeking indemnification as they
are incurred, provided the director executes an agreement to repay
the amount to be paid or reimbursed if there is a final
determination by a court of competent jurisdiction that such person
is not entitled to indemnification.
S-28
Future legislation or executive or private sector actions may
increase the difficulty and cost for us to commercialize our
products and affect the prices obtained for such
products.
In the
U.S., there have been and continue to be a number of legislative
initiatives to contain healthcare costs. For example, in March
2010, the Patient Protection and Affordable Care Act (the
“PPACA”), was enacted, which substantially changed the
way healthcare is financed by both governmental and private
insurers, and significantly impacted the U.S. pharmaceutical
industry.
Some of
the provisions of the ACA have yet to be fully implemented, and
there have been legal and political challenges to certain aspects
of the ACA. Since January 2017, President Trump has signed two
executive orders and other directives designed to delay,
circumvent, or loosen certain requirements mandated by the ACA.
Concurrently, Congress has considered legislation that would repeal
or repeal and replace all or part of the ACA. While Congress has
not passed comprehensive repeal legislation, two bills affecting
the implementation of certain taxes under the ACA have been signed
into law. The Tax Cuts and Jobs Act of 2017 (the “Tax
Act”), includes a provision that repealed, effective January
1, 2019, the tax-based shared responsibility payment imposed by the
ACA on certain individuals who fail to maintain qualifying
healthcare coverage for all or part of a year, that is commonly
referred to as the “individual mandate.” Additionally,
on January 22, 2018, President Trump signed a continuing resolution
on appropriations for fiscal year 2018 that delayed the
implementation of certain ACA-mandated fees, including the
so-called “Cadillac” tax on certain high cost
employer-sponsored insurance plans, the annual fee imposed on
certain health insurance providers based on market share, and the
medical device excise tax on non-exempt medical devices. Further,
the Bipartisan Budget Act of 2018 (“BBA”), among other
things, amended the ACA, effective January 1, 2019, to close the
coverage gap in most Medicare drug plans, commonly referred to as
the “donut hole.” In July 2018, Centers for Medicare
& Medicaid Services (“CMS”) published a final rule
permitting further collections and payments to and from certain
ACA- qualified healthcare plans and healthcare insurance issuers
under the ACA risk adjustment program in response to the outcome of
federal district court litigation regarding the method CMS uses to
determine this risk adjustment. On December 14, 2018, a U.S.
District Court Judge in the Northern District of Texas ruled that
the individual mandate is an inseverable feature of the ACA, and
therefore, because it was repealed as part of the Tax Act, the
remaining provisions of the ACA are invalid as well. While the
Texas U.S. District Court Judge, as well as the Trump
Administration and CMS have stated that the ruling will have no
immediate effect, it is unclear how this decision, subsequent
appeals, and other efforts to repeal and replace the ACA will
impact the ACA and our business.
The
increasing cost of healthcare as a percentage of GDP and the
massive and increasing deferred liabilities behind most
governmental healthcare programs (such as Medicare and Medicaid and
state and local healthcare programs especially for retirement
benefits) continue to be an economic challenge which threatens the
overall economic health of the U.S. High cost healthcare products
and therapies that are early in their life cycle are attractive
targets for parties that believe that the cost of healthcare must
be better controlled and significantly reduced. Pharmaceutical
prices and healthcare reform have been debated and acted upon by
legislators for many years. Future legislation or executive or
private sector actions related to healthcare reform could
materially and adversely affect our business by reducing our
ability to generate revenue at prices sufficient to reward for the
risks and costs of pharmaceutical development, to raise capital,
and to market our products.
There
is no assurance that federal or state healthcare reform will not
adversely affect our future business and financial results, and we
cannot predict how future federal or state legislative, judicial or
administrative changes relating to healthcare reform and
third-party payers will affect the pharmaceutical industry in
general, and our business in particular.
Even if we are able to commercialize any drug candidate, such drug
candidate may become subject to unfavorable pricing regulations or
third-party coverage and reimbursement policies, which would harm
our business.
Our
ability to commercialize any products successfully will depend, in
part, on the extent to which coverage and adequate reimbursement
for these products and related treatments will be available from
third-party payors, such as government authorities, private
healthcare insurers and health maintenance organizations. Patients
who are prescribed medications for the treatment of their
conditions generally rely on third-party payors to reimburse all or
part of the costs associated with their prescription drugs.
Coverage and adequate reimbursement from government healthcare
programs, such as Medicare and Medicaid, and private healthcare
insurers are critical to new product acceptance. Patients are
unlikely to use our future products, if any, unless coverage is
provided and reimbursement is adequate to cover a significant
portion of the cost.
S-29
Cost-containment is
a priority in the U.S. healthcare industry and elsewhere. As a
result, government authorities and other third-party payors have
attempted to control costs by limiting coverage and the amount of
reimbursement for particular medications. Increasingly, third-party
payors are requiring that drug companies provide them with
predetermined discounts from list prices and are challenging the
prices charged for medical products. Third-party payors also may
request additional clinical evidence beyond the data required to
obtain marketing approval, requiring a company to conduct expensive
pharmacoeconomic studies in order to demonstrate the medical
necessity and cost- effectiveness of its products. Commercial
third-party payors often rely upon Medicare coverage policy and
payment limitations in setting their reimbursement rates, but also
have their own methods and approval process apart from Medicare
determinations. Therefore, coverage and reimbursement for
pharmaceutical products in the U.S. can differ significantly from
payor to payor. We cannot be sure that coverage and adequate
reimbursement will be available for any product that we
commercialize and, if reimbursement is available, that the level of
reimbursement will be adequate. Coverage and reimbursement may
impact the demand for, or the price of, any drug candidate for
which we obtain marketing approval. If coverage and reimbursement
are not available or are available only at limited levels, we may
not be able to successfully commercialize any drug candidate for
which we obtain marketing approval.
Additionally, the
regulations that govern regulatory approvals, pricing and
reimbursement for new drugs and therapeutic biologics vary widely
from country to country. Some countries require approval of the
sale price of a drug or therapeutic biologic before it can be
marketed. In many countries, the pricing review period begins after
marketing approval is granted. In some foreign markets,
prescription pharmaceutical pricing remains subject to continuing
governmental control even after initial approval is granted. As a
result, we might obtain regulatory approval for a product in a
particular country, but then be subject to price regulations that
delay our commercial launch of the product, possibly for lengthy
time periods, and negatively impact the revenues we are able to
generate from the sale of the product in that country. Adverse
pricing limitations may hinder our ability to recoup our investment
in one or more drug candidates, even if our drug candidates obtain
regulatory approval.
Politically divided governmental actions and related political
actions outside of government can impact the FDA’s role in
the timely and effective review of new pharmaceutical products in
the U.S. and our business may be adversely impacted.
A
relevant example of dysfunctional government was the 35-day
government shutdown that ended February 15, 2019 which limited the
FDA to activities necessary to address imminent threats to human
life and to activities funded by carry-over user fees. Future
government shutdowns or other activities which limit the financial
resources available to the FDA (and in particular to the Center for
Drug Evaluation and Research) will delay the processing of new
product drug development submissions, reviews, and approvals and
other required regulatory actions. Such delays will adversely
impact our business and financial condition.
Effective collaboration with the FDA’s Center for Drug
Evaluation and Research (“CDER”) for the approval of
drug candidates is a highly demanding process which can result in
increased time and expense to gain approvals.
Our
lead drug development program, Validive, will be reviewed by CDER.
Efficient and professional collaboration with the FDA’s CDER
is essential for the timely clinical testing, test evaluations,
analysis and approval of our drug candidates. CDER has an
outstanding record of drug approvals and substantial funds to
operate a highly professional organization, but is also very
demanding as to the quality of clinical research and applications
for marketing approvals for drug candidates.
Our
Company has in-house expertise and experience in the management of
drug approvals. Qualified consultants and drug research
organizations are also available to aid in our drug approval
process; however, there is a meaningful risk that discussions and
interactions inherent in the drug approval process and future
developments or new improvements will result in delays, added
expenses and new scientific/medical requirements which will cause
adverse financial results and will likely impact the price of the
Company’s stock.
Future tax reform measures may negatively impact our financial
position.
Tax
reform measures are unpredictable and can change as the U.S.
congress and executive leadership changes. For example, on December
22, 2017, the Tax Cuts and Jobs Act of 2017 was signed into law
that significantly revised the Internal Revenue Code of 1986, as
amended (the “Code”). It is difficult to predict what
future tax reform measures, if any, could be implemented and the
extent to which they will impact our financial condition and our
business.
Foreign currency exchange rates may adversely affect our
consolidated financial statements.
Sales
and purchases in currencies other than the U.S. Dollar expose us to
fluctuations in foreign currencies relative to the U.S. Dollar and
may adversely affect our consolidated financial statements.
Increased strength of the U.S. Dollar increases the effective price
of our future drug products sold in U.S. Dollars into other
countries, which may require us to lower our prices or adversely
affect sales to the extent we do not increase local currency
prices. Decreased strength of the U.S. Dollar could adversely
affect the cost of materials, products and services we purchase
overseas. Sales and expenses of our non-U.S. businesses are also
translated into U.S. Dollars for reporting purposes and the
strengthening or weakening of the U.S. Dollar could result in
unfavorable foreign currency translation and transaction effects.
In addition, certain of our businesses may in the future invoice
customers in a currency other than the business’ functional
currency, and movements in the invoiced currency relative to the
functional currency could also result in unfavorable foreign
currency translation and transaction effects. We also face exchange
rate risk from our investments in subsidiaries owned and operated
in foreign countries.
S-30
Our anticipated operating expenses and capital expenditures over
the next year are based upon our management’s estimates of
possible future events. Actual amounts and the cost of new
conditions could differ materially from those estimated by our
management.
Development of
pharmaceuticals and cancer drugs is extremely risky and
unpredictable. We have estimated operating expenses and capital
expenditures over the next year based on certain assumptions. Any
change in the assumptions could and will cause the actual results
to vary substantially from the anticipated expenses and
expenditures and could result in material differences in actual
versus forecasted expenses or expenditures. Furthermore, all of the
factors are subject to the effect of unforeseeable future events.
The estimates of capital expenditures and operating expenses
represent forward-looking statements within the meaning of the
federal securities laws. Prospective investors are cautioned that
any such forward-looking statements are not guarantees of future
performance and involve risks and uncertainties. Actual events or
results may differ materially from those discussed in the
forward-looking statements as a result of various factors,
including the risk factors set forth under this “Risk
Factors” section in this prospectus supplement. In view of
the foregoing, investors should not rely on these estimates in
making a decision to invest in us.
The financial and operational projections that we may make from
time to time are subject to inherent risks.
The
projections that we provide herein or our management may provide
from time to time (including, but not limited to, the cost and
timing of our Phase 3 clinical trials, clinical and regulatory
timelines, production and supply matters, commercial launch dates,
and other financial or operational matters) reflect numerous
assumptions made by our management, including assumptions with
respect to our specific as well as general business, regulatory,
economic, market and financial conditions and other matters, all of
which are difficult to predict and many of which are beyond our
control. Accordingly, there is a risk that the assumptions made in
preparing the projections, or the projections themselves, will
prove inaccurate. There may be differences between actual and
projected results, and actual results may be materially different
from those contained in the projections. The inclusion of the
projections in this prospectus should not be regarded as an
indication that we, our management, the underwriters or their
respective representatives considered or consider the projections
to be a guaranteed prediction of future events, and the projections
should not be relied upon as such. See “Cautionary Statement
Concerning Forward-Looking Statements.”
Our present and potential future international operations may
expose us to business, political, operational, and financial risks
associated with doing business outside of the United
States.
Our
business is subject to risks associated with conducting business
internationally. Some of our suppliers and clinical research
organizations and clinical trial sites are located outside of the
U.S. Furthermore, if we or any future collaborator succeeds in
developing any products, we anticipate marketing them in the EU and
other jurisdictions in addition to the U.S. If approved, we or our
collaborator may hire sales representatives and conduct physician
and patient association outreach activities outside of the U.S.
Doing business internationally involves a number of risks,
including but not limited to:
●
multiple,
conflicting and changing laws and regulations such as privacy
regulations, tax laws, export and import restrictions, employment
laws, regulatory requirements, and other governmental approvals,
permits and licenses;
●
failure by us
to obtain and maintain regulatory approvals for the use of our
products in various countries;
●
rejection or
qualification of foreign clinical trial data by the competent
authorities of other countries;
●
additional
potentially relevant third-party patent and other intellectual
property rights that may be necessary to develop and commercialize
our products and drug candidates;
●
complexities
and difficulties in obtaining, maintaining, enforcing and defending
our patent and other intellectual property rights;
●
difficulties
in staffing and managing foreign operations;
●
complexities
associated with managing multiple payor reimbursement regimes,
government payors or patient self-pay systems;
●
limits in our
ability to penetrate international markets;
●
financial
risks, such as longer payment cycles, difficulty collecting
accounts receivable, the impact of local and regional financial
crises on demand and payment for our products and exposure to
foreign currency exchange rate fluctuations;
●
natural
disasters, political and economic instability, including wars,
terrorism and political unrest, outbreak of disease, boycotts,
curtailment of trade and other business restrictions,
implementation of tariffs;
●
certain
expenses including, among others, expenses for travel, translation
and insurance; and
●
regulatory
and compliance risks that relate to anti-corruption compliance and
record-keeping that may fall within the purview of the U.S. Foreign
Corrupt Practices Act, its accounting provisions or its
anti-bribery provisions or provisions of anti-corruption or
anti-bribery laws in other countries.
Any of
these factors could harm our ongoing international clinical
operations and supply chain, as well as any future international
expansion and operations and, consequently, our business, financial
condition, prospects and results of operations.
S-31
Our future growth may depend, in part, on our ability to operate in
foreign markets, where we would be subject to additional regulatory
burdens and other risks and uncertainties.
Our
future growth may depend, in part, on our ability to develop and
commercialize drug candidates in foreign markets for which we may
rely on partnering with third parties. We will not be permitted to
market or promote any drug candidate before we receive regulatory
approval from the applicable regulatory authority in a foreign
market, and we may never receive such regulatory approval for any
drug candidate. To obtain separate regulatory approval in foreign
countries, we generally must comply with numerous and varying
regulatory requirements of such countries regarding safety and
efficacy and governing, among other things, clinical trials and
commercial sales, pricing and distribution of a drug candidate, and
we cannot predict success in these jurisdictions. If we obtain
approval of any of our current or potential future drug candidates
and ultimately commercialize any such drug candidate in foreign
markets, we would be subject to risks and uncertainties, including
the burden of complying with complex and changing foreign
regulatory, tax, accounting and legal requirements and the reduced
protection of intellectual property rights in some foreign
countries.
We are subject to U.S. and foreign anti-corruption and anti-money
laundering laws with respect to our operations and non-compliance
with such laws can subject us to criminal or civil liability and
harm our business.
We are
subject to the U.S. Foreign Corrupt Practices Act of 1977, as
amended (“the FCPA”), the U.S. domestic bribery statute
contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA
PATRIOT Act, and possibly other state and national anti-bribery and
anti-money laundering laws in countries in which we conduct
activities. Anti-corruption laws are interpreted broadly and
prohibit companies and their employees, agents, third-party
intermediaries, joint venture partners and collaborators from
authorizing, promising, offering or providing, directly or
indirectly, improper payments or benefits to recipients in the
public or private sector. We interact with officials and employees
of government agencies and government-affiliated hospitals,
universities and other organizations. In addition, we may engage
third-party intermediaries to promote our clinical research
activities abroad or to obtain necessary permits, licenses and
other regulatory approvals. We can be held liable for the corrupt
or other illegal activities of these third-party intermediaries,
our employees, representatives, contractors, partners and agents,
even if we do not explicitly authorize or have actual knowledge of
such activities.
We have
a Code of Business Conduct and Ethics which mandates compliance
with the FCPA and other anti-corruption laws applicable to our
business throughout the world. However, we cannot assure you that
our employees and third-party intermediaries will comply with this
code or such anti-corruption laws. Noncompliance with
anti-corruption and anti-money laundering laws could subject us to
whistleblower complaints, investigations, sanctions, settlements,
prosecution, other enforcement actions, disgorgement of profits,
significant fines, damages, other civil and criminal penalties or
injunctions, suspension or debarment from contracting with certain
persons, the loss of export privileges, reputational harm, adverse
media coverage and other collateral consequences. If any subpoenas,
investigations or other enforcement actions are launched, or
governmental or other sanctions are imposed, or if we do not
prevail in any possible civil or criminal litigation, our business,
results of operations and financial condition could be materially
harmed. In addition, responding to any action will likely result in
a materially significant diversion of management’s attention
and resources and significant defense and compliance costs and
other professional fees. In certain cases, enforcement authorities
may even cause us to appoint an independent compliance monitor
which can result in added costs and administrative
burdens.
S-32
FORWARD-LOOKING STATEMENTS
This
prospectus contains “forward-looking statements” within
the meaning of Section 27A of the Securities Act of 1933, as
amended (the “Act”) and Section 21E of the 34 Act. All
statements other than statements of historical facts included in
this Prospectus are forward-looking statements. The words
“hopes,” “believes,”
“anticipates,” “plans,”
“seeks,” “estimates,”
“projects,” “expects,”
“intends,” “may,” “could,”
“should,” “would,” “will,”
“continue,” and similar expressions are intended to
identify forward-looking statements. The following uncertainties
and factors, among others, could affect future performance and
cause actual results to differ materially from those matters
expressed in or implied by forward-looking statements:
●
our ability to
raise sufficient funds in the coming months in order for us to
start our Validive Phase 3 clinical trial;
●
our ability to find
a suitable pharmaceutical partner to further our development of
Validive, if we are unable to raise sufficient additional
financing;
●
risks and
uncertainties associated with our research and development
activities, including our clinical trials;
●
estimated
timeframes for our clinical trials and regulatory reviews for
approval to market products;
●
plans to research,
develop and commercialize our current and future product
candidates;
●
the rate and degree
of market acceptance and clinical utility of any products for which
we receive marketing approval;
●
commercialization,
marketing and manufacturing capabilities and strategy;
●
intellectual
property position and strategy;
●
future financial
performance;
●
estimates regarding
expenses, capital requirements and need for additional
financing;
●
the impact of
government laws and regulations;
●
ability to attract
and retain key personnel; and
●
financial and
operational projections.
Although we believe
that the expectations reflected in such forward-looking statements
are appropriate, we can give no assurance that such expectations
will be realized. All subsequent written and oral forward-looking
statements attributable to us or persons acting on our behalf are
expressly qualified in their entirety by the cautionary statements
above and made elsewhere in this prospectus and future supplemental
prospectuses. We undertake no obligation to update any statements
made in this Prospectus or elsewhere, including without limitation
any forward-looking statements, except as required by
law.
You
should read this prospectus and the documents that we reference in
this prospectus with the understanding that our actual future
results, levels of activity, performance and events and
circumstances may be materially different from what we
expect.
S-33
The
amount of proceeds from this offering will depend upon the number
of shares of our Common Stock sold and the market price at which
they are sold. There can be no assurance that we will be able to
sell any shares under or fully utilize the sales agreement with the
Agent as a source of financing.
We
intend to use the net proceeds of this offering for our operations,
including, but not limited to, general corporate purposes, which
may include research and development expenditures, clinical trial
expenditures, manufacture and supply of product and working
capital. The precise amount, use, and timing of the application of
such proceeds will depend upon our funding requirements and the
availability and cost of other capital. Pending application of the
net proceeds as described above, we intend to invest the net
proceeds of the offering in short- term, investment-grade,
interest-bearing securities and/or savings accounts.
We
will need to raise significant additional funds or find a suitable
pharmaceutical partner to start the Validive clinical trial
program, to complete it and take Validive through potential
approval and, if approved, through commercialization, to support
further development of camsirubicin, MNPR-101 and any additional
product candidates.
S-34
Our
historical net tangible book value as of September 30, 2019, was
$4.6 million, or $0.50 per share of our Common Stock.
After
giving effect to the sale of our Common Stock in the aggregate
amount of $19.7 million at an assumed price of $22.00 per share,
the last reported sale price of our Common Stock on Nasdaq on
January 10, 2020, and after deducting estimated offering
commissions and expenses payable by us, after giving effect to the
issuance of 18,433 shares of common stock pursuant to a stock
option exercise on November 25, 2019 for $109,998 and our issuance
and sale of 1,277,778 shares of our Common Stock in our initial
public offering price at $8.00 per share, after deducting
underwriting discounts and commissions and estimated offering
expenses payable by us, our as adjusted net tangible book value as
of September 30, 2019, would have been $33.1 million, or $2.88 per
share. This represents an immediate increase in net tangible book
value per share of $2.38 to existing stockholders and immediate
dilution of $19.12 in net tangible book value per share to new
investors purchasing our Common Stock in this
offering.
Dilution per share
to new investors is determined by subtracting as adjusted net
tangible book value per share after this offering from the assumed
price per share paid by new investors. The following table
illustrates this dilution on a per share basis.
|
Assumed
Price Per Share
|
|
$ 22.00
|
|
Historical Net
Tangible Book Value Per Share as of September 30, 2019
|
$ 0.50
|
|
|
Increase in Net
Tangible Book Value Per Share Attributable to New
Investors
|
2.38
|
|
|
As
Adjusted Net Tangible Book Value Per Share After this
Offering
|
|
2.88
|
|
Dilution
Per Share to New Investors
|
|
$ 19.12
|
The
foregoing table is based on an aggregate 10,587,632 shares of our
Common Stock outstanding as of January 10, 2020 and
excludes:
●
1,096,463 shares of
our Common Stock issuable upon the exercise of outstanding stock
options (weighted-average exercise price of $3.06; 758,685 shares
vested); and
●
485,104 shares of
our Common Stock reserved for issuance under our 2016 Stock
Incentive Plan.
Except as otherwise indicated, all information in this prospectus
assumes:
●
All shares
referenced above and throughout this prospectus take into account a
70-for-1 stock split of our Common Stock effected in March
2017.
All of
the foregoing is illustrative only. There can be no assurance we
will sell all, or any, of the shares offered by this prospectus,
when any such sales will occur or what the sales prices will
be.
S-35
We have
never declared or paid cash dividends on our capital stock. We
currently intend to retain all available funds and future earnings,
if any, for use in the operation of our business and do not
anticipate paying any cash dividends on our Common Stock in the
foreseeable future. Any future determination to declare and pay
dividends will be made at the discretion of our Board and will
depend on various factors, including applicable laws, our results
of operations, our financial condition, our capital requirements,
general business conditions, our future prospects and other factors
that our Board may deem relevant. Additionally, our ability to pay
dividends on our capital stock could be limited by terms and
covenants of any future indebtedness. Investors should not purchase
our Common Stock with the expectation of receiving cash
dividends.
S-36
On
January 13, 2020 we entered into a Sales Agreement (the
“Sales Agreement”) with JonesTrading Institutional
Services LLC (the “Agent”) under which we may issue and
sell our Common Stock having an aggregate gross sales price of up
to $19,700,000 from time to time through or to the Agent, acting as
agent or principal. A copy of the Sales Agreement is filed as an
exhibit herein.
Each time that we wish to issue and sell shares of
our Common Stock under the Sales Agreement, if any, we will provide
the Agent with a placement notice describing the amount of shares
to be sold or the gross proceeds to be raised in a given time
period, the time period during which sales are requested to be
made, any limitation on the amount of shares of Common Stock that
may be sold in any single day, any minimum price below which sales
may not be made or any minimum price requested for sales in a given
time period and any other instructions relevant to such requested
sales. Upon delivery of a placement notice and subject to
the terms and conditions of the Sales Agreement, the Agent may sell
our Common Stock by any method permitted by law deemed to be
“at the market offerings” as defined in Rule 415 under
the Securities Act. We may instruct the Agent not to sell our
Common Stock if the sales cannot be effected at or above the price
designated by us from time to time. We or the Agent may suspend the
offering of our Common Stock upon notice and subject to other
conditions.
We will
pay the Agent commissions, in cash, for its services in acting as
an agent in the sale of our Common Stock. The Agent will be
entitled to compensation of up to 3.0% of the gross proceeds from
each sale of our Common Stock. Because there is no minimum offering
amount required as a condition to this offering, the actual total
public offering amount, commissions and proceeds to us, if any, are
not determinable at this time. We have also agreed to reimburse the
Agent for certain specified expenses, including the fees and
disbursements of their legal counsel in an amount not to exceed
$40,000. Additionally, pursuant to the terms of the sales
agreement, we agreed to reimburse
the Agent for the documented fees and costs
of its legal counsel reasonably incurred in connection with the
Agent’s ongoing diligence in an amount not to exceed $5,000
per year. We estimate that the total expenses for the offering,
excluding compensation and reimbursements payable to the Agent
under the terms of the Sales Agreement, will be approximately
$40,000.
Settlement for
sales of our Common Stock will occur on the second business day
following the date on which any sales are made, or on some other
date that is agreed upon by us and the Agent in connection with a
particular transaction, in return for payment of the net proceeds
to us. Sales of our Common Stock as contemplated in this prospectus
will be settled through the facilities of The Depository Trust
Company or by such other means as we and the Agent may agree upon.
There is no arrangement for funds to be received in an escrow,
trust or similar arrangement.
The
Agent will use their commercially reasonable best efforts,
consistent with its sales and trading practices, to solicit offers
to purchase our Common Stock under the terms and subject to the
conditions set forth in the Sales Agreement. In connection with the
sale of our Common Stock on our behalf, the Agent will be deemed to
be an “underwriter” within the meaning of the
Securities Act and the compensation of the Agent will be deemed to
be an underwriting commission or discount. We have agreed to
provide indemnification and contribution to the Agent against
certain civil liabilities, including liabilities under the
Securities Act.
The
offering of our Common Stock pursuant to the Sales Agreement will
terminate upon the termination of the Sales Agreement as permitted
therein. We and the Agent may terminate the Sales Agreement at any
time upon five days’ prior notice or by the Agent at any time
in certain circumstances, including the occurrence of a material
and adverse change in our business or financial condition that
makes it impractical or inadvisable to market our Common Stock or
to enforce contracts for the sale of our Common Stock.
The
Agent and its respective affiliates have in the past and may in the
future provide various investment banking, commercial banking and
other financial services for us, for which services they may in the
future receive customary fees.
This
prospectus in electronic format may be made available on a website
maintained by the Agent who may distribute this prospectus
electronically.
S-37
Certain
legal matters will be passed upon for us by Baker & Hostetler,
LLP, Columbus, Ohio. Certain legal matters in connection with this
offering will be passed upon for the Agent by Duane Morris LLP, New
York, New York.
EXPERTS
The
financial statements as of December 31, 2018 and 2017, and for the
years then ended, included in this Prospectus have been so included
in reliance on the report of BPM LLP, an independent registered
public accounting firm, given on the authority of said firm as
experts in auditing and accounting.
WHERE
YOU CAN FIND MORE INFORMATION
We have
filed with the SEC a shelf registration statement on Form S-3 under
the Securities Act with respect to the Common Stock we are offering
by this prospectus. This prospectus does not contain all of the
information included in the registration statement. For further
information pertaining to us and our Common Stock, you should refer
to the registration statement and to its exhibits. Whenever we make
reference in this prospectus to any of our contracts, agreements or
other documents, the references are not necessarily complete, and
you should refer to the exhibits attached to the registration
statement for copies of the actual contract, agreement or other
document.
We file
annual, quarterly and current reports, information statements and
proxy statements and other information with the SEC. You can read
our SEC filings, including the registration statement, at the
SEC’s website at www.sec.gov. You may also read and copy any
document we file with the SEC at its public reference facility at
100 F Street, N.E., Room 1580, Washington, D.C. 20549. We also
maintain a website at http://www.monopartx.com. You may access,
free of charge, our annual reports on Form 10-K, quarterly reports
on Form 10- Q, current reports on Form 8-K and amendments to those
reports filed or furnished pursuant to Section 13(a) or 15(d) of
the Exchange Act as soon as reasonably practicable after such
material is electronically filed with, or furnished to, the SEC.
The information contained on, or that can be accessed through, our
website is not a part of, and should not be construed as being
incorporated by reference into, this prospectus or the accompanying
prospectus supplement.
You may
also obtain copies of the documents at prescribed rates by writing
to the Public Reference Section of the SEC at 100 F Street, N.E.,
Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for
further information on the operation of the public reference
facilities.
S-38
INCORPORATION OF DOCUMENTS BY
REFERENCE
The SEC
allows us to incorporate by reference the information we file with
it, which means that we can disclose important information to you
by referring you to another document that we have filed separately
with the SEC. You should read the information incorporated by
reference because it is an important part of this prospectus.
Information in this prospectus supersedes information incorporated
by reference that we filed with the SEC prior to the date of this
prospectus, while information that we file later with the SEC will
automatically update and supersede the information in this
prospectus. We incorporate by reference into this prospectus and
the registration statement of which this prospectus is a part the
information and documents listed below that we have filed with the
SEC:
●
our Quarterly
Report on Form
10-Q for the
quarter ended September 30, 2019, filed with the SEC on November
12, 2019;
●
our Quarterly
Report on Form
10-Q for the
quarter ended June 30, 2019, filed with the SEC on August 8,
2019;
●
our Quarterly
Report on Form
10-Q for the
quarter ended March 31, 2019, filed with the SEC on May 10,
2019;
●
our Information
Statement regarding our Annual Meeting of Stockholders on June 27,
2019, on DEF14C, filed
with the SEC on May 22, 2019;
●
our Annual Report
on Form
10-K for the year
ended December 31, 2018, filed with the SEC on February 26,
2019;
●
our Current Reports
on Form 8-K, filed with the SEC on January 7, 2020 and1 June 27, 2019,
to the extent the information in such reports is filed and not
furnished; and
●
the description of
our Common Stock contained in our Registration Statement
on Form 8-A,
registering our Common Stock under Section 12(b) under the Exchange
Act, filed with the SEC on September 30, 2019, as supplemented by
the "Description of Capital Stock" beginning on page 5 of this
prospectus and including any amendments or reports filed for the
purpose of updating such description.
We also
incorporate by reference any future filings (other than Current
Reports furnished under Item 2.02 or Item 7.01 of Form 8-K and
exhibits filed on such form that are related to such items unless
such Form 8-K expressly provides to the contrary) made with the SEC
pursuant to Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act
until we file a post-effective amendment that indicates the
termination of the offering of the common stock made by this
prospectus and will become a part of this prospectus from the date
that such documents are filed with the SEC. Information in such
future filings updates and supplements the information provided in
this prospectus. Any statements in any such future filings will
automatically be deemed to modify and supersede any information in
any document we previously filed with the SEC that is incorporated
or deemed to be incorporated herein by reference to the extent that
statements in the later filed document modify or replace such
earlier statements.
We will
furnish without charge to each person, including any beneficial
owner, to whom a prospectus is delivered, upon written or oral
request, a copy of any or all of the documents incorporated by
reference into this prospectus but not delivered with the
prospectus, including exhibits that are specifically incorporated
by reference into such documents. You should direct any requests
for documents to Monopar Therapeutics, Inc., Attention: Corporate
Secretary, 1000 Skokie Blvd., Suite 350, Wilmette, IL 60091. Our
phone number is (847)
388-0349. You may also view the documents that we file with the SEC
and incorporate by reference in this Prospectus on our corporate
website at www.monopartx.com. The information on our website is not
incorporated by reference and is not a part of this
prospectus.
S-39
PROSPECTUS
$75,000,000
Common Stock
We
may offer and sell an indeterminate number of shares of our Common
Stock from time to time under this prospectus. You should read this
prospectus and any prospectus supplement carefully before you
invest.
We
may offer our Common Stock in one or more offerings in amounts, at
prices, and on terms determined at the time of the offering. We may
sell our Common Stock through agents we select or through
underwriters and dealers we select. If we use agents, underwriters
or dealers, we will name them and describe their compensation in a
prospectus supplement.
This
prospectus provides a general description of our Common Stock that
we may offer. Each time we sell our Common Stock, we will provide
specific terms of the Securities offered in a supplement to this
prospectus. The prospectus supplement may also add, update or
change information contained in this prospectus. You should read
this prospectus and the applicable prospectus supplement carefully
before you invest in our Common Stock. This prospectus may not be
used to consummate a sale of our Common Stock unless accompanied by
the applicable prospectus supplement.
Our
Common Stock is listed for trading on the Nasdaq Capital Market
under the symbol “MNPR.” On January 2, 2020 the last
reported sale price of our Common Stock was $17.27 per share. As of
that date, and based on that price, the aggregate market value of
our voting and non-voting common equity held by non-affiliates was
approximately $37.9 million.
We have not offered and sold any securities pursuant to General
Instruction I.B.6 of Form S-3 during the 12 calendar months prior
to and including the date of this prospectus.
Investing in our Common Stock involves significant risks. See
“Risk Factors” included in any accompanying prospectus
supplement and in the documents incorporated by reference in this
prospectus for a discussion of the factors you should carefully
consider before deciding to purchase our Common Stock.
Neither the Securities and Exchange Commission nor any state
securities commission has approved or disapproved of these
securities or passed upon the accuracy or adequacy of this
prospectus. Any representation to the contrary is a criminal
offense.
The
date of this Prospectus is January 13, 2020
TABLE
OF CONTENTS
i
We are a clinical stage biopharmaceutical company focused on
developing proprietary therapeutics designed to improve clinical
outcomes for cancer patients. We are building a drug development
pipeline through the licensing and acquisition of oncology
therapeutics in late preclinical and clinical development stages.
We leverage our scientific and clinical experience to help reduce
the risk and accelerate the clinical development of our drug
product candidates. We currently have three compounds in
development: Validive® (clonidine
mucobuccal tablet; clonidine MBT), a Phase 3-ready, first-in-class
mucoadhesive buccal tablet for the prevention and treatment of
radiation-induced severe oral mucositis in oropharyngeal cancer
patients; camsirubicin (generic name for
5-imino-13-deoxydoxorubicin; previously known as MNPR-201,
GPX-150), a proprietary Phase 2 clinical stage topoisomerase
II-alpha selective analog of doxorubicin engineered specifically to
retain anticancer activity while minimizing toxic effects on the
heart; and MNPR-101 (formerly huATN-658), a pre-IND stage humanized
monoclonal antibody, which targets the urokinase plasminogen
activator receptor (“uPAR”), for the treatment of
advanced solid cancers.
Our
principal executive offices are located at 1000 Skokie Blvd, Suite
350, Wilmette, IL 60091. Our telephone number is (847)
388-0349.
You should consider carefully
the risks discussed under the section captioned “Risk
Factors” contained in our annual report on Form 10-K for the
year ended December 31, 2018 and in our subsequent quarterly
reports on Form 10-Q, as updated by our subsequent
filings
under the Securities Exchange Act of 1934, as amended (the
“Exchange Act”), each of which is incorporated by
reference in this prospectus in its entirety, together with other
information in this prospectus, and the information and documents
incorporated by reference in this prospectus, any prospectus
supplement and any free writing prospectus that we have authorized
for use in connection with this offering before you make a decision
to invest in our securities. If any of these events actually occur,
our business, operating results, prospects
or financial condition could be materially and adversely affected.
This could cause the trading price of our common stock to decline
and you may lose all or part of your investment.
IMPORTANT INFORMATION ABOUT THIS
PROSPECTUS
In
this prospectus, unless the context suggests otherwise, references
to “Monopar Therapeutics,” “Monopar,” the
“Company,” “we,” “us” and
“our” refer to Monopar Therapeutics Inc.
This prospectus is part of a “shelf”
registration statement. By using a shelf registration statement, we
may sell our Common Stock, as described in this prospectus, from
time to time in one or more offerings. Each time we sell our Common
Stock, we will provide a prospectus supplement to this prospectus
that contains specific information about the terms of such
offering. The prospectus supplement may also add, update or change
information contained in this prospectus. Before purchasing our
Common Stock, you should carefully read both this prospectus and
any prospectus supplement, together with the additional information
incorporated into this prospectus or described under the heading
“Where You Can Find More
Information.”
You
should rely only on the information contained or incorporated by
reference in this prospectus and any prospectus supplement. We have
not authorized any other person to provide you with different
information. If anyone provides you with different or inconsistent
information, you should not rely on it. We will not make an offer
to sell our Common Stock in any jurisdiction where the offer or
sale is not permitted. You should assume that the information
appearing in this prospectus, and have incorporated by reference,
is accurate only as of the date on the front cover of this
prospectus, or when such document was filed with the Securities and
Exchange Commission (“SEC”). Our business, financial
condition, results of operations and prospects may have changed
since the relevant date.
Neither
we, nor any of our officers, directors, agents, representatives or
underwriters, make any representation to you about the legality of
an investment. You should not interpret the contents of this
prospectus, any prospectus supplement, or any free writing
prospectus to be legal, business, investment or tax advice. You
should consult with your own advisors for that type of advice and
consult with them about the legal, tax, business, financial and
other issues that you should consider before investing in our
Common Stock.
We
will not use this prospectus to offer and sell our Common Stock
unless it is accompanied by a prospectus supplement that more fully
describes the terms of the offering.
FORWARD-LOOKING STATEMENTS
This Prospectus
contains “forward-looking statements” within the
meaning of Section 27A of the Securities Act of 1933, as amended
(the “Act”) and Section 21E of the 34 Act. All
statements other than statements of historical facts included in
this Prospectus are forward-looking statements. The words
“hopes,” “believes,”
“anticipates,” “plans,”
“seeks,” “estimates,”
“projects,” “expects,”
“intends,” “may,” “could,”
“should,” “would,” “will,”
“continue,” and similar expressions are intended to
identify forward-looking statements. The following uncertainties
and factors, among others, could affect future performance and
cause actual results to differ materially from those matters
expressed in or implied by forward-looking statements:
●
our
ability to raise sufficient funds in the coming months in order for
us to start our Validive Phase 3 clinical trial;
●
our
ability to find a suitable pharmaceutical partner to further our
development of Validive, if we are unable to raise sufficient
additional financing;
●
risks
and uncertainties associated with our research and development
activities, including our clinical trials;
●
estimated
timeframes for our clinical trials and regulatory reviews for
approval to market products;
●
plans
to research, develop and commercialize our current and future
product candidates;
●
the
rate and degree of market acceptance and clinical utility of any
products for which we receive marketing approval;
●
commercialization,
marketing and manufacturing capabilities and strategy;
●
intellectual
property position and strategy;
●
future
financial performance;
●
estimates regarding
expenses, capital requirements and need for additional
financing;
●
the
impact of government laws and regulations;
●
ability
to attract and retain key personnel; and
●
financial and
operational projections.
Although we believe
that the expectations reflected in such forward-looking statements
are appropriate, we can give no assurance that such expectations
will be realized. All subsequent written and oral forward-looking
statements attributable to us or persons acting on our behalf are
expressly qualified in their entirety by the cautionary statements
above and made elsewhere in this prospectus and future supplemental
prospectuses. We undertake no obligation to update any statements
made in this Prospectus or elsewhere, including without limitation
any forward-looking statements, except as required by
law.
You should read
this prospectus and the documents that we reference in this
prospectus with the understanding that our actual future results,
levels of activity, performance and events and circumstances may be
materially different from what we expect.
DESCRIPTION OF CAPITAL STOCK
We have the
authority to issue 40,000,000 shares of Common Stock, $0.001 par
value. As of January 3, 2020, there were 10,587,632 shares of our
Common Stock issued and outstanding.
We have reserved
1,600,000 shares of our Common Stock for issuance under our 2016
Stock Incentive Plan, as subsequently amended (the
“Plan”), and as of January 3, 2020, we have outstanding
stock options to purchase up to 1,087,463 shares of our Common
Stock and 494,104 shares of our Common Stock available for future
stock awards under the Plan.
Common
Stock
Voting Rights
The holders of
shares of our Common Stock are entitled to one vote per share for
the election of directors and on all other matters submitted to a
vote of stockholders. Shares of our Common Stock do not have
cumulative voting rights. The election of our Board of Directors
(“Board”) is decided by a plurality of the votes cast
at a meeting of our stockholders by the holders of stock entitled
to vote in the election.
Dividends
Holders of our
Common Stock are entitled to receive such dividends as may be
declared by our Board out of funds legally available
therefor.
Liquidation
Upon our
dissolution and liquidation, holders of our Common Stock are
entitled to a ratable share of our net assets remaining after
payments to our creditors.
Rights and Preferences
Our stockholders have
no preemptive rights to acquire additional shares of our Common
Stock or other securities. The shares of our Common Stock are not
subject to redemption.
Preferred
Stock
We have no
preferred stock authorized or outstanding.
Anti-Takeover
Provisions
Delaware Law
We are subject to
Section 203 of the Delaware General Corporation Law. Subject to
certain exceptions, Section 203 prevents a publicly held Delaware
corporation from engaging in a "business combination" with any
"interested stockholder" for three years following the date that
the person became an interested stockholder, unless the interested
stockholder attained such status with the approval of our Board or
unless the business combination is approved in a prescribed manner.
A "business combination" includes, among other things, a merger or
consolidation involving us and the "interested stockholder" and the
sale of more than 10% of our assets. In general, an "interested
stockholder" is any entity or person beneficially owning 15% or
more of our outstanding voting stock and any entity or person
affiliated with or controlling or controlled by such entity or
person.
Authorized but Unissued Shares
The authorized but
unissued shares of our Common Stock are available for future
issuance without stockholder approval, subject to any limitations
imposed by the listing standards of any exchange on which our
shares are listed. These additional shares may be used for a
variety of corporate finance transactions, acquisitions and
employee benefit plans. The existence of authorized but unissued
and unreserved Common Stock could make more difficult or discourage
an attempt to obtain control of us by means of a proxy contest,
tender offer, merger or otherwise.
Election of Director by Plurality of Shares; Vacancies
Our
Amended and Restated By-laws provide that directors will be elected
by a plurality of votes cast by the shares present in person or by
proxy at a meeting of the stockholders and entitled to vote
thereon, a quorum being present at such meeting. There is no
cumulative voting, meaning that Directors may be elected with a
vote of holders of less than a majority of the outstanding common
stock.
Our Amended and
Restated By-laws also provide that vacancies occurring on our Board
may be filled by the affirmative votes of a majority of the
remaining members of our Board or by the sole remaining director,
and not by our stockholders. Such provisions in our corporate
organizational documents and under Delaware law may prevent or
frustrate attempts by our stockholders to change our management or
hinder efforts to acquire a controlling interest in us. The
inability to make changes to our Board could prevent or discourage
an attempt to take control of the Company by means of a proxy
contest, tender offer, merger or otherwise.
Special Meeting of Stockholders; Advance Notice Requirements for
Stockholder Proposals and Director Nominations; Stockholder
Action
Our Amended and
Restated By-laws provide that, except as otherwise required by law,
special meetings of the stockholders can only be called by our
Board. Stockholders at a special meeting may only consider matters
set forth in the notice of the meeting. These provisions could have
the effect of delaying until the next stockholder meeting
stockholder actions that may be favored by the holders of a
majority of our outstanding voting securities.
Super Majority Voting
The General
Corporation Law of the State of Delaware provides generally that
the affirmative vote of a majority of the shares entitled to vote
on any matter is required to amend a corporation's certificate of
incorporation or by-laws, unless a corporation's certificate of
incorporation or by-laws, as the case may be, requires a greater
percentage. Our Amended and Restated By-laws may be amended or
repealed by a majority vote of our Board or the affirmative vote of
the holders of at least a majority of the votes that all our
stockholders would be entitled to cast in any election of
Directors.
Registration
Rights
We are subject to
an agreement with TacticGem, LLC (“TacticGem”), our
largest stockholder, which obligates us to file a Form S-3 or other
appropriate form of registration statement covering the resale of
any of our Common Stock by TacticGem, or its members Gem
Pharmaceuticals, LLC, or Tactic Pharma, LLC, upon direction by
TacticGem at any time after we have been subject to the reporting
requirements of the 1934 Act for at least twelve months (the
“Initial Holding Period”). We are required to use our
best efforts to have such registration statement declared effective
as soon as practical after it is filed. In the event that such
registration statement for resale is not approved by the SEC, and
TacticGem submits a written request, we are required to prepare and
file a registration statement on Form S-1 registering such Common
Stock for resale and to use our best efforts to have such
registration statement declared effective as soon as practical
thereafter. After registration, pursuant to these rights, these
shares will become freely tradable without restriction under the
Securities Act other than pursuant to restrictions on affiliates
under Rule 144. TacticGem has agreed to enter into a lock-up
agreement and to not exercise any rights of resale for 180 days
after the date of our initial public offering which was December
18, 2019.
Listing
Our Common Stock is
listed on the Nasdaq Capital Market under the symbol
“MNPR.”
Transfer
Agent and Registrar
The transfer
agent and registrar for our Common Stock is VStock Transfer, LLC
(“VStock”). VStock’s address is 18 Lafayette
Place, Woodmere, NY 11598
We
may sell our Common Stock covered in this prospectus in any of
three ways (or in any combination):
●
through
underwriters or dealers;
●
directly to a
limited number of purchasers or to a single purchaser;
or
The distribution of
our Common Stock may be effected from time to time in one or more
transactions:
●
at market prices
prevailing at the time of sale;
●
at prices related
to such prevailing market prices; or
Each time that we
use this prospectus to sell our Common Stock, we will also provide
a prospectus supplement that contains the specific terms of the
offering. The prospectus supplement will set forth the terms of the
offering of our Common Stock, including:
●
the name or names
of any underwriters, dealers or agents and the amounts of any of
our Common Stock underwritten or purchased by each of them;
and
●
the public offering
price of our Common Stock and the proceeds to us and any discounts,
commissions or concessions allowed or reallowed or paid to
dealers.
Any public offering
price and any discounts or concessions allowed or reallowed or paid
to dealers may be changed from time to time.
If underwriters are
used in the sale of our Common Stock, our Common Stock will be
acquired by the underwriters for their own account and may be
resold from time to time in one or more transactions, including
negotiated transactions, at a fixed public offering price or at
varying prices determined at the time of sale. Our Common Stock may
be either offered to the public through underwriting syndicates
represented by managing underwriters, or directly by underwriters.
Generally, the underwriters’ obligations to purchase our
Common Stock will be subject to certain conditions precedent. The
underwriters may be obligated to purchase all of our Common Stock
if they purchase any of our Common Stock.
We may sell the
securities through agents from time to time. The prospectus
supplement will name any agent involved in the offer or sale of our
Common Stock and any commissions we pay to them. Generally, any
agent will be acting on a best efforts basis for the period of its
appointment.
We may authorize
underwriters, dealers or agents to solicit offers by certain
purchasers to purchase our Common Stock from us at the public
offering price set forth in the prospectus supplement pursuant to
delayed delivery contracts providing for payment and delivery on a
specified date in the future. The contracts will be subject only to
those conditions set forth in the prospectus supplement, and the
prospectus supplement will set forth any commissions we pay for
solicitation of these contracts.
Agents and
underwriters may be entitled to indemnification by us against
certain civil liabilities, including liabilities under the
Securities Act of 1933, as amended, or to contribution with respect
to payments which the agents or underwriters may be required to
make in respect thereof. Agents and underwriters may be customers
of, engage in transactions with, or perform services for us in the
ordinary course of business.
We may enter into
derivative transactions with third parties, or sell our Common
Stock not covered by this prospectus to third parties in privately
negotiated transactions. If the applicable prospectus supplement
indicates, in connection with those derivatives, the third parties
may sell our Common Stock covered by this prospectus and the
applicable prospectus supplement, including in short sale
transactions. If so, the third party may use our Common Stock
pledged by us or borrowed from us or others to settle those sales
or to close out any related open borrowings of our Common Stock,
and may use our Common Stock received from us in settlement of
those derivatives to close out any related open borrowings of our
Common Stock. The third party in such sale transactions will be an
underwriter and will be identified in the applicable prospectus
supplement (or a post-effective amendment).
Certain legal
matters will be passed upon for us by Baker & Hostetler, LLP,
Columbus, Ohio.
The financial
statements as of December 31, 2018 and 2017, and for the years then
ended, included in this Prospectus have been so included in
reliance on the report of BPM LLP, an independent registered public
accounting firm, given on the authority of said firm as experts in
auditing and accounting.
WHERE
YOU CAN FIND MORE INFORMATION
We have filed with
the SEC this shelf registration statement on Form S-3 under the
Securities Act with respect to our Common Stock we are offering by
this prospectus. This prospectus does not contain all of the
information included in the registration statement. For further
information pertaining to us and our Common Stock, you should refer
to the registration statement and to its exhibits. Whenever we make
reference in this prospectus to any of our contracts, agreements or
other documents, the references are not necessarily complete, and
you should refer to the exhibits attached to the registration
statement for copies of the actual contract, agreement or other
document.
We file annual,
quarterly and current reports, information statements and proxy
statements and other information with the SEC. You can read our SEC
filings, including the registration statement, at the SEC’s
website at www.sec.gov. You may also read and copy any document we
file with the SEC at its public reference facility at 100 F Street,
N.E., Room 1580, Washington, D.C. 20549. We also maintain a website
at http://www.monopartx.com. You may access, free of charge, our
annual reports on Form 10-K, quarterly reports on Form 10- Q,
current reports on Form 8-K and amendments to those reports filed
or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act
as soon as reasonably practicable after such material is
electronically filed with, or furnished to, the SEC. The
information contained on, or that can be accessed through, our
website is not a part of, and should not be construed as being
incorporated by reference into, this prospectus or the accompanying
prospectus supplement.
You may also obtain copies of the documents at prescribed rates by
writing to the Public Reference Section of the SEC at 100 F Street,
N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330
for further information on the operation of the public reference
facilities.
INCORPORATION OF DOCUMENTS BY REFERENCE
The SEC allows us
to incorporate by reference the information we file with it, which
means that we can disclose important information to you by
referring you to another document that we have filed separately
with the SEC. You should read the information incorporated by
reference because it is an important part of this prospectus.
Information in this prospectus supersedes information incorporated
by reference that we filed with the SEC prior to the date of this
prospectus, while information that we file later with the SEC will
automatically update and supersede the information in this
prospectus. We incorporate by reference into this prospectus and
the registration statement of which this prospectus is a part the
information and documents listed below that we have filed with the
SEC (Commission File No. 000-55866):
●
our
Quarterly Report on
Form 10-Q for the quarter ended September 30, 2019, filed with
the SEC on November 12, 2019;
●
our
Quarterly Report on
Form 10-Q for the quarter ended June 30, 2019, filed with the
SEC on August 8, 2019;
●
our
Quarterly Report on
Form 10-Q for the quarter ended March 31, 2019, filed with the
SEC on May 10, 2019;
●
our
Information Statement regarding our Annual Meeting of Stockholders
on June 27, 2019, on
DEF14C, filed with the SEC on May 22, 2019;
●
our
Annual Report on
Form 10-K for the year ended December 31, 2018, filed with the
SEC on February 26, 2019;
●
the
description of our Common Stock contained in our Registration
Statement on
Form 8-A, registering our Common Stock under Section 12(b)
under the Exchange Act, filed with the SEC on September 30, 2019,
as supplemented by the "Description of Capital Stock" beginning on
page 5 of this prospectus and including any amendments or reports
filed for the purpose of updating such
description.
We also incorporate
by reference any future filings (other than Current Reports
furnished under Item 2.02 or Item 7.01 of Form 8-K and exhibits
filed on such form that are related to such items unless such Form
8-K expressly provides to the contrary) made with the SEC pursuant
to Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act,
including those made after the date of the initial filing of the
registration statement of which this prospectus is a part and prior
to effectiveness of such registration statement, until we file a
post-effective amendment that indicates the termination of the
offering of the common stock made by this prospectus and will
become a part of this prospectus from the date that such documents
are filed with the SEC. Information in such future filings updates
and supplements the information provided in this prospectus. Any
statements in any such future filings will automatically be deemed
to modify and supersede any information in any document we
previously filed with the SEC that is incorporated or deemed to be
incorporated herein by reference to the extent that statements in
the later filed document modify or replace such earlier
statements.
We will furnish
without charge to each person, including any beneficial owner, to
whom a prospectus is delivered, upon written or oral request, a
copy of any or all of the documents incorporated by reference into
this prospectus but not delivered with the prospectus, including
exhibits that are specifically incorporated by reference into such
documents. You should direct any requests for documents to Monopar
Therapeutics, Inc., Attention: Corporate Secretary, 1000 Skokie
Blvd., Suite 350, Wilmette, IL 60091. Our phone number is
(847) 388-0349. You may also
view the documents that we file with the SEC and incorporate by
reference in this Prospectus on our corporate website at
www.monopartx.com. The information on our website is not
incorporated by reference and is not a part of this
prospectus.
$19,700,000

Common Stock
PROSPECTUS SUPPLEMENT
January 13,
2020