UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
Quarterly Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the Quarterly Period Ended
Transition Report Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the transition period from to
Commission File Number:
(Exact name of registrant as specified in its charter) |
| ||
(State or other jurisdiction of incorporation or organization) |
| (I.R.S. employer identification number) |
|
|
|
| ||
(Address of principal executive offices) |
| (zip code) |
(
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
| Trading Symbol(s) |
| Name of each exchange on which registered |
|
| The (Nasdaq Capital Market) |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ | Accelerated filer | ☐ |
☒ | Smaller reporting company | ||
|
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes
The number of shares outstanding with respect to each of the classes of our common stock, as of October 31, 2021, is set forth below:
Class | Number of shares outstanding | |
Common Stock, par value $0.001 per share |
MONOPAR THERAPEUTICS INC.
TABLE OF CONTENTS
| 2 |
Forward-Looking Statements
This Quarterly Report on Form 10-Q contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Act”), and Section 21E of the Securities Exchange Act of 1934, as amended. All statements other than statements of historical facts included in this Quarterly Report on Form 10-Q are forward-looking statements. The words “hopes,” “believes,” “anticipates,” “plans,” “seeks,” “estimates,” “projects,” “expects,” “intends,” “may,” “could,” “should,” “would,” “will,” “continue,” and similar expressions are intended to identify forward-looking statements. The following uncertainties and factors, among others, could affect future performance and cause actual results to differ materially from those matters expressed in or implied by forward-looking statements:
● | our ability to raise sufficient funds in the next 12 months in order for us to complete the Phase 3 portion of our Validive Phase 2b/3 clinical trial and, if required, complete a second confirmatory Phase 3 clinical trial, to continue the clinical development of camsirubicin beyond the Phase 1b dose escalation clinical trial, to support further development of potential MNPR-101-derived radioimmunotherapeutics (RITs) and companion diagnostics to treat severe COVID-19 (patients with SARS-CoV-2 infection) and cancer, to support further development of MNPR-101, MNPR-202 and related compounds and generally to support our current and any future product candidates through completion of clinical trials, approval processes and, if applicable, commercialization; |
● | our ability to find a suitable pharmaceutical partner or partners to further our development efforts, if we are unable to raise sufficient additional financing; |
● | risks and uncertainties associated with our research and development activities, including our clinical trials; |
● | estimated timeframes for our clinical trials and regulatory reviews for approval to market products; |
● | the rate of market acceptance, relative market price compared to alternative therapies, and the competitive clinical efficacy and safety of any products for which we receive marketing approval; |
● | the difficulties of commercialization, marketing and product manufacturing and overall strategy; |
● | uncertainties of intellectual property position and strategy; |
● | delivering strong future financial performance; |
● | our ability to attract and retain key personnel and to find and utilize external sources of experience, expertise and scientific, medical and commercialization knowledge to complete product development and commercialization of new products; |
● | the risks inherent in our estimates regarding the level of needed expenses, capital requirements and the availability of required additional financing; |
● | the impact of government laws and regulations including increased governmental control of healthcare, including direct price controls driving lower prices and other governmental regulations affecting cost requirements and structures required to deliver therapeutic products; |
● | the uncertain impact of the COVID-19 pandemic on our ability to advance our clinical programs and raise additional financing; and |
● | uncertainty of our financial and operational projections and the development of new competitive products and technologies. |
Although we believe that the expectations reflected in such forward-looking statements are appropriate, we can give no assurance that such expectations will be realized. Cautionary statements are disclosed in this Quarterly Report on Form 10-Q. All subsequent written and oral forward-looking statements attributable to us or persons acting on our behalf are expressly qualified in their entirety by the cautionary statements. We undertake no obligation to update any statements made in this Quarterly Report on Form 10-Q or elsewhere, including without limitation any forward-looking statements, except as required by law.
Any forward-looking statements in this Quarterly Report on Form 10-Q reflect our current views with respect to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements. Information that is based on estimates, forecasts, projections, market research or similar methodologies is inherently subject to uncertainties and actual events or circumstances may differ materially from events and circumstances projected in this information.
| 3 |
| Table of Contents |
Risks Associated With Our Business
Our business is subject to numerous risks and uncertainties, including those highlighted in “Item 1A - Risk Factors” of our Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 25, 2021. These risks include, among others, the following:
● | We are a clinical stage biopharmaceutical company with a history of financial losses. We expect to continue to incur significant losses for the foreseeable future and may never achieve or maintain cash self-sufficiency or profitability, which could result in a decline in the market value of our common stock. |
● | Funds raised to date are not sufficient to 1) complete our Validive Phase 2b/3 ("VOICE") clinical program, including, if required, completing a second Phase 3 confirmatory clinical trial; 2) continue the clinical development of camsirubicin beyond the Phase 1b dose escalation clinical trial; 3) to support further development of potential MNPR-101-derived radioimmunotherapeutics and companion diagnostics to treat severe COVID-19 (patients with SARS-CoV-2 infection) and cancer; or 4) to support continued development of MNPR-101, MNPR-202 and related compounds. If we are unable to raise enough funds in the next 12 months from the sale of our common stock or other financing efforts, we will have to consider strategic options such as out-licensing Validive or other product candidates, entering into a clinical or commercial partnership, or terminating one or more programs. There can be no assurance that we can find a suitable partner on satisfactory terms. |
● | We have a limited operating history, no revenues from operations, and are dependent upon raising substantial capital to continue our drug development programs. |
● | We do not have and may never have any approved products on the market. Our business is highly dependent upon receiving approvals from various U.S. and international governmental agencies and will be severely harmed if we are not granted approval to manufacture and sell our product candidates. |
● | Our clinical trials may not yield sufficiently conclusive results for regulatory agencies to approve the use of our products, which will adversely affect our financial condition. |
● | If we experience delays or difficulties in the enrollment of subjects in clinical trials, our receipt of necessary regulatory approvals will be delayed or prevented, which will materially delay our program schedules and adversely affect our financial condition. |
● | We rely on third parties to conduct our drug product manufacturing, non-clinical studies, and our clinical trials. If these third parties do not or cannot successfully carry out their contractual duties or meet expected deadlines or performance goals, the initiation or conduct of our clinical trials may be delayed and we may be unable to obtain regulatory approval for, or commercialize, our current product candidates or any future products, and our financial condition will be adversely affected. |
● | We face significant competition from other biotechnology and pharmaceutical companies, in targeted medical indications, and our operating results will suffer if we fail to compete effectively. Competition and technological change may make our product candidates obsolete or non-competitive. |
● | The termination of third-party licenses will adversely affect our rights to important compounds or technologies. |
● | If we and our third-party licensors do not obtain and preserve protection for our respective intellectual property rights, our competitors may be able to develop competing drugs, which will adversely affect our financial condition. |
● | If we lose key management leadership, and/or scientific personnel, and if we cannot recruit qualified employees or other highly qualified and experienced significant personnel for future personnel requirements, we may experience program delays and increased compensation and operational costs, and our business will be materially disrupted. |
● | The ongoing COVID-19 pandemic could have a substantial negative impact on our business, financial condition, operating results, stock price and ability raise additional funds. |
| 4 |
| Table of Contents |
PART I
FINANCIAL INFORMATION
Item 1. Financial Statements
Monopar Therapeutics Inc.
Condensed Consolidated
Balance Sheets
(Unaudited)
|
| September 30, 2021 |
|
| December 31, 2020* |
| ||
Assets |
|
|
|
|
|
| ||
Current assets: |
|
|
|
|
|
| ||
Cash and cash equivalents |
| $ |
|
| $ |
| ||
Other current assets |
|
|
|
|
|
| ||
Total current assets |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Other non-current assets |
|
|
|
|
|
| ||
Total assets |
| $ |
|
| $ |
| ||
Liabilities and Stockholders' Equity |
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Accounts payable, accrued expenses and other current liabilities |
| $ |
|
| $ |
| ||
Total current liabilities and total liabilities |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
Commitments and contingencies (Note 6) |
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
|
Common stock, par value of $ |
|
|
|
|
|
| ||
Additional paid-in capital |
|
|
|
|
|
| ||
Accumulated other comprehensive loss |
|
| ( | ) |
|
| ( | ) |
Accumulated deficit |
|
| ( | ) |
|
| ( | ) |
Total stockholders' equity |
|
|
|
|
|
| ||
Total liabilities and stockholders' equity |
| $ |
|
| $ |
| ||
* Derived from the Company’s audited consolidated financial statements.
The accompanying notes are an integral part of these condensed consolidated financial statements.
| 5 |
| Table of Contents |
Monopar Therapeutics Inc.
Condensed Consolidated
Statements of Operations and Comprehensive Loss
(Unaudited)
|
| Three Months Ended September 30, |
|
| Nine Months Ended September 30, |
| ||||||||||
|
| 2021 |
|
| 2020 |
|
| 2021 |
|
| 2020 |
| ||||
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Research and development |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
General and administrative |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Total operating expenses |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Loss from operations |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Other income: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Net loss |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Other comprehensive income: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Foreign currency translation gain |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Comprehensive loss |
| $ | ( | ) |
| $ | ( | ) |
| $ | ( | ) |
| $ | ( | ) |
Net loss per share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted |
| $ | ( | ) |
| $ | ( | ) |
| $ | ( | ) |
| $ | ( | ) |
Weighted average shares outstanding: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted |
|
|
|
|
|
|
|
|
|
|
|
| ||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| 6 |
| Table of Contents |
Monopar Therapeutics Inc.
Condensed Consolidated Statements of Stockholders’ Equity
Nine Months Ended September 30, 2021
(Unaudited)
|
| Common Stock |
|
| Additional Paid-in |
|
| Accumulated Other Comprehensive |
|
| Accumulated |
|
| Total Stockholders' |
| |||||||||
|
| Shares |
|
| Amount |
|
| Capital |
|
| Loss |
|
| Deficit |
|
| Equity |
| ||||||
Balance at January 1, 2021 |
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ | ( | ) |
| $ |
| ||||
Issuance of common stock under a Capital on DemandTM Sales Agreement with JonesTrading Institutional Services LLC, net of commissions and fees of $338,153 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Issuance of common stock to non-employee directors pursuant to vested restricted stock units |
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
| |||||
Issuance of common stock to employees pursuant to vested restricted stock units, net of taxes |
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
| ( | ) | ||||
Issuance of common stock upon exercise of stock options |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Stock-based compensation (non-cash) |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Net loss |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | |||
Other comprehensive income |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Balance at March 31, 2021 |
|
|
|
|
|
|
|
| ( | ) |
| ( | ) |
|
| |||||||||
Issuance of common stock to non-employee directors pursuant to vested restricted stock units |
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
| |||||
Issuance of common stock to employees pursuant to vested restricted stock units, net of taxes |
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
| ( | ) | ||||
Stock-based compensation (non-cash) |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Net loss |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | |||
Other comprehensive loss |
|
|
|
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
| ( | ) | |||
Balance at June 30, 2021 |
|
|
|
|
|
|
|
| ( | ) |
| ( | ) |
|
| |||||||||
Issuance of common stock to non-employee directors pursuant to vested restricted stock units |
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
| |||||
Issuance of common stock to employees pursuant to vested restricted stock units, net of taxes |
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
| ( | ) | ||||
Stock-based compensation (non-cash) |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Net loss |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | |||
Other comprehensive income |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Balance at September 30, 2021 |
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ | ( | ) |
| $ |
| ||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| 7 |
| Table of Contents |
Monopar Therapeutics Inc.
Condensed Consolidated Statements of Stockholders’ Equity
Nine Months Ended September 30, 2020
(Unaudited)
|
| Common Stock |
|
| Additional Paid-in |
|
| Accumulated Other Comprehensive |
|
| Accumulated |
|
| Total Stockholders' |
| |||||||||
|
| Shares |
|
| Amount |
|
| Capital |
|
| Loss |
|
| Deficit |
|
| Equity |
| ||||||
Balance at January 1, 2020 |
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ | ( | ) |
| $ |
| ||||
Issuance of common stock under a Capital on DemandTM Sales Agreement with JonesTrading Institutional Services LLC, net of commissions and fees of $16,284 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Issuance of common stock to non-employee directors pursuant to vested restricted stock units |
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
| |||||
Stock-based compensation (non-cash) |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Offering costs |
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
| ( | ) | |||
Net loss |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | |||
Other comprehensive loss |
|
| — |
|
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
| ( | ) | |||
Balance at March 31, 2020 |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
|
| ||||
Issuance of common stock under a Capital on DemandTM Sales Agreement with JonesTrading Institutional Services LLC, net of commissions and fees of $29,425 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Issuance of common stock to non-employee directors pursuant to vested restricted stock units |
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
|
| ||||
Stock-based compensation (non-cash) |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Offering costs |
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
|
| ( | ) | |
Net loss |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | |||
Other comprehensive income |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Balance at June 30, 2020 |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
|
| ||||
Issuance of common stock under a Capital on DemandTM Sales Agreement with JonesTrading Institutional Services LLC, net of commissions and fees of $207,326 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Issuance of common stock to non-employee directors pursuant to vested restricted stock units |
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
| |||||
Stock-based compensation (non-cash) |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Offering costs |
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
|
| ( | ) | |
Net loss |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | |||
Other comprehensive income |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Balance at September 30, 2020 |
|
|
|
| $ |
|
| $ |
|
| $ | ( | ) |
| $ | ( | ) |
| $ |
| ||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| 8 |
| Table of Contents |
Monopar Therapeutics Inc.
Condensed Consolidated
Statements of Cash Flows
(Unaudited)
|
| For the Nine Months Ended September 30, |
| |||||
|
| 2021 |
|
| 2020 |
| ||
Cash flows from operating activities: |
|
|
|
|
|
| ||
Net loss |
| $ | ( | ) |
| $ | ( | ) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
Stock-based compensation expense (non-cash) |
|
|
|
|
|
| ||
Changes in operating assets and liabilities, net |
|
|
|
|
|
|
|
|
Other current assets |
|
| ( | ) |
|
| ( | ) |
Accounts payable, accrued expenses and other current liabilities |
|
|
|
|
| ( | ) | |
Net cash used in operating activities |
|
| ( | ) |
|
| ( | ) |
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
Cash proceeds from the sales of common stock under a Capital on DemandTM Sales Agreement with JonesTrading Institutional Services LLC, net of commissions, fees and offering costs of $338,153 and $253,035 for the nine months ended September 30, 2021, and 2020, respectively |
|
|
|
|
|
| ||
Other offering costs |
|
|
|
|
| ( | ) | |
Taxes paid related to net share settlement of vested restricted stock units |
|
| ( | ) |
|
|
| |
Cash proceeds from the issuance of stock upon exercise of stock options |
|
|
|
|
|
| ||
PPP forgivable bank loan |
|
|
|
|
|
| ||
Net cash provided by financing activities |
|
|
|
|
|
| ||
Effect of exchange rates |
|
|
|
|
|
| ||
Net increase (decrease) in cash and cash equivalents |
|
|
|
|
|
| ||
Cash and cash equivalents at beginning of period |
|
|
|
|
|
| ||
Cash and cash equivalents at end of period |
| $ |
|
| $ |
| ||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| 9 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
Note 1 - Nature of Business and Liquidity
Nature of Business
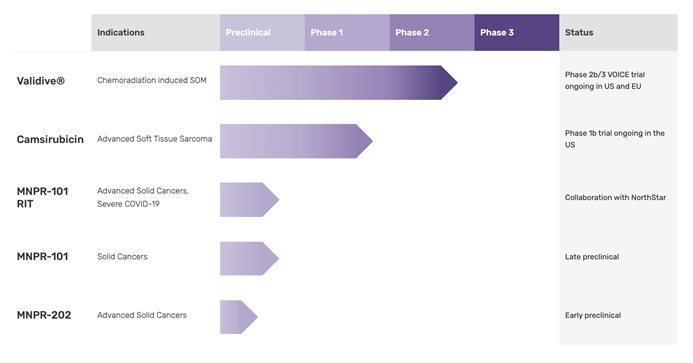
Monopar Therapeutics Inc. (“Monopar” or the “Company”) is a clinical-stage biopharmaceutical company focused on developing proprietary therapeutics designed to extend life or improve quality of life for cancer patients. Monopar currently has four compounds in development: 1) Validive® (clonidine hydrochloride mucobuccal tablet; clonidine HCI MBT), a Phase 2b/3 clinical stage, first-in-class mucoadhesive buccal tablet for the prevention of radiation induced severe oral mucositis (“SOM”) in oropharyngeal cancer patients; 2) camsirubicin (generic name for MNPR-201, GPX-150; 5-imino-13-deoxydoxorubicin), a Phase 1b clinical stage novel analog of doxorubicin engineered specifically to retain anticancer activity while minimizing toxic effects on the heart; 3) MNPR-101, a preclinical stage uPAR-targeted antibody being developed as a radioimmunotherapeutic and companion diagnostic for advanced cancers and severe COVID-19; and 4) an early stage camsirubicin analog, MNPR-202, for various cancers.
Liquidity
The Company has incurred an accumulated deficit of approximately $
The coronavirus disease (“COVID-19”) pandemic continues to affect economies and business around the world. In response to COVID-19 and its effects on clinical trials, in 2020 Monopar modified the original adaptive design Phase 3 clinical trial for its lead product candidate, Validive, to be a Phase 2b/3 clinical trial (“VOICE”) to better fit the types of trials which can enroll sufficient required patients in the current environment. This modification allowed the Company to activate the VOICE clinical trial without requiring near-term financing. To complete the VOICE clinical program, including, if required, completing a second Phase 3 confirmatory clinical trial, Monopar will require additional funding in the millions or tens of millions of dollars (depending on if the Company has consummated a collaboration or partnership or neither for Validive), which it is planning to pursue in the next 12 months. Due to many uncertainties, the Company is unable to estimate the pandemic’s financial impact or duration in light of vaccine rollouts and adverse new cases surging from COVID-19 variants at this time, or its potential impact on the Company’s current clinical trials, including COVID-19’s effect on drug candidate manufacturing, shipping, patient recruitment at clinical sites and regulatory agencies around the globe.
Note 2 - Significant Accounting Policies
Basis of Presentation
These condensed consolidated financial statements include the financial results of Monopar Therapeutics Inc., its wholly-owned French subsidiary, Monopar Therapeutics, SARL, and its wholly-owned Australian subsidiary, Monopar Therapeutics Australia Pty Ltd and have been prepared in accordance with accounting principles generally accepted in the U.S. (“GAAP”) and include all disclosures required by GAAP for financial reporting. All intercompany accounts have been eliminated. The principal accounting policies applied in the preparation of these condensed consolidated financial statements are set out below and have been consistently applied in all periods presented. The Company has been primarily involved in performing research activities, developing product candidates, and raising capital to support and expand these activities.
| 10 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
The accompanying unaudited condensed consolidated financial statements contain all normal, recurring adjustments necessary to present fairly the Company’s condensed consolidated financial position as of September 30, 2021, the Company’s condensed consolidated results of operations and comprehensive loss for the three and nine months ended September 30, 2021, and 2020, and the Company’s condensed consolidated cash flows for the nine months ended September 30, 2021, and 2020.
The condensed consolidated results of operations and comprehensive loss and condensed consolidated cash flows for the periods presented are not necessarily indicative of the consolidated results of operations or cash flows which may be reported for the remainder of 2021 or for any future period. Certain information and footnote disclosures normally included in financial statements prepared in accordance with GAAP have been condensed or omitted. The accompanying unaudited interim condensed consolidated financial statements should be read in conjunction with the audited financial statements and notes thereto for the year ended December 31, 2020, included in the Company’s Annual Report on Form 10-K filed with the U.S. Securities and Exchange Commission (the “SEC”) on March 25, 2021.
Functional Currency
The Company's consolidated functional currency is the U.S. Dollar. The Company's Australian subsidiary and French subsidiary use the Australian Dollar and European Euro, respectively, as their functional currency. At each quarter-end, each foreign subsidiary's balance sheets are translated into U.S. Dollars based upon the quarter-end exchange rate, while their statements of operations and comprehensive loss and statements of cash flows are translated into U.S. Dollars based upon an average exchange rate during the period.
Comprehensive Loss
Comprehensive loss represents net loss plus any gains or losses not reported in the condensed consolidated statements of operations and comprehensive loss, such as foreign currency translations gains and losses that are typically reflected on the Company’s condensed consolidated statements of stockholders’ equity.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities, and reported amounts of expenses in the condensed consolidated financial statements and accompanying notes. Actual results could differ from those estimates.
Going Concern Assessment
The Company applies Accounting Standards Codification 205-40 (“ASC 205-40”), Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern, which the Financial Accounting Standards Board (“FASB”) issued to provide guidance on determining when and how reporting companies must disclose going concern uncertainties in their financial statements. ASC 205-40 requires management to perform interim and annual assessments of an entity’s ability to continue as a going concern within one year of the date of issuance of the entity’s financial statements (or within one year after the date on which the financial statements are available to be issued, when applicable). Further, a company must provide certain disclosures if there is “substantial doubt about the entity’s ability to continue as a going concern.” In October 2021, the Company analyzed its cash requirements through December 2022 and has determined that, based upon the Company’s current available cash, the Company has no substantial doubt about its ability to continue as a going concern.
| 11 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
Cash Equivalents
The Company considers all highly liquid investments purchased with a maturity of 90 days or less on the date of purchase to be cash equivalents. Cash equivalents as of September 30, 2021, and December 31, 2020, consisted of one money market account.
Deferred Offering Costs
Deferred offering costs represent legal, auditing, travel and filing fees related to fundraising efforts that have not yet been concluded. There were no deferred offering costs on the Balance Sheets as of September 30, 2021, and December 31, 2020
Prepaid Expenses
Prepayments are expenditures for goods or services before the goods are used or the services are received and are charged to operations as the benefits are realized. Prepaid expenses may include payments to development collaborators in excess of actual expenses incurred by the collaborator, measured at the end of each reporting period. Prepayments also include insurance premiums, dues and subscriptions and software costs of $
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentration of credit risk consist of cash and cash equivalents. The Company maintains cash and cash equivalents at two reputable financial institutions. As of September 30, 2021, the balance at one financial institution was in excess of the $
Fair Value of Financial Instruments
For financial instruments consisting of cash and cash equivalents, accounts payable, accrued expenses, and other current liabilities, the carrying amounts are reasonable estimates of fair value due to their relatively short maturities.
The Company adopted ASC 820, Fair Value Measurements and Disclosures, as amended, which addresses the measurement of the fair value of financial assets and financial liabilities. Under this standard, fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (i.e., the “exit price”) in an orderly transaction between market participants at the measurement date.
The standard establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs reflect assumptions market participants would use in pricing an asset or liability based on market data obtained from independent sources. Unobservable inputs reflect a reporting entity’s pricing an asset or liability developed based on the best information available under the circumstances. The fair value hierarchy consists of the following three levels:
Level 1 - instrument valuations are obtained from real-time quotes for transactions in active exchange markets involving identical assets.
Level 2 - instrument valuations are obtained from readily available pricing sources for comparable instruments.
Level 3 - instrument valuations are obtained without observable market values and require a high-level of judgment to determine the fair value.
| 12 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
Determining which category an asset or liability falls within the hierarchy requires significant judgment. The Company evaluates its hierarchy disclosures each reporting period. There were no transfers between Level 1, 2 or 3 of the fair value hierarchy during the three and nine months ended September 30, 2021, and 2020. The following table presents the assets and liabilities recorded that are reported at fair value on our condensed consolidated balance sheets on a recurring basis. No values were recorded in Level 2 or Level 3 at September 30, 2021, and December 31, 2020.
Assets and Liabilities Measured at Fair Value on a Recurring Basis
September 30, 2021 |
| Level 1 |
|
| Total |
| ||
Assets |
|
|
|
|
|
| ||
Cash equivalents(1) |
| $ |
|
| $ |
| ||
Total |
| $ |
|
| $ |
| ||
December 31, 2020 |
| Level 1 |
|
| Total |
| ||
Assets |
|
|
|
|
|
| ||
Cash equivalents(1) |
| $ |
|
| $ |
| ||
Total |
| $ |
|
| $ |
| ||
(1) | Cash equivalents represent the fair value of the Company’s investment in a money market account. |
Net Loss per Share
Net loss per share for the three and nine months ended September 30, 2021, and 2020 is calculated by dividing net loss by the weighted-average shares of common stock outstanding during the period. Diluted net loss per share for the three and nine months ended September 30, 2021, and 2020 is calculated by dividing net loss by the weighted-average shares of the sum of a) weighted average common stock outstanding (
Research and Development Expenses
Research and development (“R&D”) costs are expensed as incurred. Major components of R&D expenses include salaries and benefits paid to the Company’s R&D staff, compensation expenses of G&A personnel performing R&D, fees paid to consultants and to the entities that conduct certain R&D activities on the Company’s behalf and materials and supplies which were used in R&D activities during the reporting period.
| 13 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
Clinical Trial Expense
The Company accrues and expenses the costs for clinical trial activities performed by third parties based upon estimates of the percentage of work completed over the life of the individual study in accordance with agreements established with contract research organizations, service providers, and clinical trial sites. The Company estimates the amounts to accrue based upon discussions with internal clinical personnel and external service providers as to progress or stage of completion of trials or services and the agreed upon fee to be paid for such services. Costs of setting up clinical trial sites for participation in the trials are expensed immediately as R&D expenses. Clinical trial site costs related to patient screening and enrollment are accrued as patients are screened/entered into the trial.
Collaborative Agreements
The Company and its collaborative partners are active participants in collaborative agreements and all parties would be exposed to significant risks and rewards depending on the technical and commercial success of the activities. Contractual payments to the other parties in collaboration agreements and costs incurred by the Company when the Company is deemed to be the principal participant for a given transaction are recognized on a gross basis in R&D expenses. Royalties and license payments are recorded as earned.
During the three and nine months ended September 30, 2021, and 2020, no milestones were met, and no royalties were earned, therefore, the Company did not pay or accrue/expense any license or royalty payments.
Licensing Agreements
The Company has various agreements licensing technology utilized in the development of its product or technology programs. The licenses contain success milestone obligations and royalties on future sales. During the three and nine months ended September 30, 2021, and 2020, no milestones were met, and no royalties were earned, therefore, the Company did not pay or accrue/expense any license or royalty payments under any of its license agreements.
Patent Costs
The Company expenses costs relating to issued patents and patent applications, including costs relating to legal, renewal and application fees, as a component of general and administrative expenses in its condensed consolidated statements of operations and comprehensive loss.
Income Taxes
The Company uses an asset and liability approach for accounting for deferred income taxes, which requires recognition of deferred income tax assets and liabilities for the expected future tax consequences of events that have been recognized in its financial statements but have not been reflected in its taxable income. Estimates and judgments are required in the calculation of certain tax liabilities and in the determination of the recoverability of certain deferred income tax assets, which arise from temporary differences and carryforwards. Deferred income tax assets and liabilities are measured using the currently enacted tax rates that apply to taxable income in effect for the years in which those tax assets and liabilities are expected to be realized or settled.
| 14 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
The Company regularly assesses the likelihood that its deferred income tax assets will be realized from recoverable income taxes or recovered from future taxable income. To the extent that the Company believes any amounts are not “more likely than not” to be realized, the Company records a valuation allowance to reduce the deferred income tax assets. In the event the Company determines that all or part of the net deferred tax assets are not realizable in the future, an adjustment to the valuation allowance would be charged to earnings in the period such determination is made. Similarly, if the Company subsequently determines deferred income tax assets that were previously determined to be unrealizable are now realizable, the respective valuation allowance would be reversed, resulting in an adjustment to earnings in the period such determination is made.
Internal Revenue Code Sections 382 and 383 (“Sections 382 and 383”) limit the use of net operating loss (“NOL”) carryforwards and R&D credits, after an ownership change. To date, the Company has not conducted a Section 382 or 383 study, however, because the Company will continue to raise significant amounts of equity in the coming years, the Company expects that Sections 382 and 383 will limit the Company’s usage of NOLs and R&D credits in the future.
ASC 740, Income Taxes, requires that the tax benefit of net operating losses, temporary differences, and credit carryforwards be recorded as an asset to the extent that management assesses that realization is "more likely than not." Realization of the future tax benefits is dependent on the Company's ability to generate sufficient taxable income within the carryforward period. The Company has reviewed the positive and negative evidence relating to the realizability of the deferred tax assets and has concluded that the deferred tax assets are not “more likely than not” to be realized. As a result, the Company recorded a full valuation allowance as of September 30, 2021, and December 31, 2020. U.S. Federal R&D tax credits from 2016 to 2019 were utilized to reduce payroll taxes in future periods and were recorded as other current assets for amounts anticipated to be received within 12 months and other non-current assets for amounts anticipated to be received beyond 12 months on the Company’s condensed consolidated balance sheets. The Company intends to maintain the valuation allowance until sufficient evidence exists to support its reversal. The Company regularly reviews its tax positions. For a tax benefit to be recognized, the related tax position must be “more likely than not” to be sustained upon examination. Any amount recognized is generally the largest benefit that is “more likely than not” to be realized upon settlement. The Company’s policy is to recognize interest and penalties related to income tax matters as an income tax expense. For the three and nine months ended September 30, 2021, and 2020, the Company did not have any interest or penalties associated with unrecognized tax benefits.
The Company is subject to U.S. Federal, Illinois and California income taxes. In addition, the Company is subject to local tax laws of France and Australia. Tax regulations within each jurisdiction are subject to the interpretation of the related tax laws and regulations and require significant judgment to apply. The Company was incorporated on December 16, 2015, and is subject to U.S. Federal, state and local tax examinations by tax authorities for the tax years 2015 through 2020. The Company does not anticipate significant changes to its current uncertain tax positions through September 30, 2021. The Company filed its U.S. Federal and state tax returns for the year ended December 31, 2020, prior to the extended filing deadlines in all jurisdictions.
Stock-Based Compensation
The Company accounts for stock-based compensation arrangements with employees, non-employee directors and consultants using a fair value method, which requires the recognition of compensation expense for costs related to all stock-based awards, including stock option and restricted stock unit (“RSU”) grants. The fair value method requires the Company to estimate the fair value of stock-based payment awards on the date of grant using an option pricing model or the closing stock price on the date of grant in the case of RSUs.
| 15 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
Stock-based compensation expense for awards granted to employees, non-employee directors and consultants are based on the fair value of the underlying instrument calculated using the Black-Scholes option-pricing model on the date of grant for stock options and using the closing stock price on the date of grant for RSUs and recognized as expense on a straight-line basis over the requisite service period, which is the vesting period. Determining the appropriate fair value model and related assumptions requires judgment, including estimating the future stock price volatility, forfeiture rates and expected terms. The expected volatility rates are estimated based on the actual volatility of comparable public companies over recent historical periods of the same length as the expected term. The Company selected these companies based on reasonably comparable characteristics, including market capitalization, stage of corporate development and with historical share price information sufficient to meet the expected term (life) of the stock-based awards. The expected term for options granted to date is estimated using the simplified method. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company has not paid dividends and does not anticipate paying a cash dividend in the future vesting period and, accordingly, uses an expected dividend yield of zero. The risk-free interest rate is based on the rate of U.S. Treasury securities with maturities consistent with the estimated expected term of the awards.
Note 3 - Capital Stock
Holders of the common stock are entitled to receive such dividends as may be declared by the Board of Directors out of funds legally available therefor. To date no dividends have been declared. Upon dissolution and liquidation of the Company, holders of the common stock are entitled to a ratable share of the net assets of the Company remaining after payments to creditors of the Company. The holders of shares of common stock are entitled to one vote per share for the election of each director nominated to the Board and one vote per share on all other matters submitted to a vote of stockholders.
The Company’s amended and restated certificate of incorporation authorizes the Company to issue
Sales of Common Stock
On January 13, 2020, the Company entered into a Capital on Demand™ Sales Agreement with JonesTrading, as sales agent, pursuant to which Monopar could offer and sell (at its discretion), from time to time, through or to JonesTrading shares of Monopar’s common stock, having an aggregate offering price of
As of September 30, 2021, the Company had
Note 4 - Stock Incentive Plan
In April 2016, the Company’s Board of Directors and stockholders representing a majority of the Company’s outstanding stock at that time, approved the Monopar Therapeutics Inc. 2016 Stock Incentive Plan, as amended (the “Plan”), allowing the Company to grant up to an aggregate
| 16 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
During the three months ended September 30, 2021, the Company’s Plan Administrator Committee (with regards to non-officer employees and consultants) and the Company’s Compensation Committee, as ratified by the Board of Directors (in the case of executive officers and non-employee directors), granted to one executive officer and one non-officer employee aggregate stock options for the purchase of
Under the Plan, the per share exercise price for the shares to be issued upon exercise of an option shall be determined by the Plan Administrator, except that the per share exercise price shall be no less than 100% of the fair market value per share on the grant date. Fair market value is the Company’s closing price on Nasdaq. Stock options generally expire after 10 years.
Stock option activity under the Plan was as follows:
|
| Options Outstanding |
| |||||
|
| Number of Shares Subject to Options |
|
| Weighted-Average Exercise Price |
| ||
Balances at January 1, 2020 |
|
|
|
| $ |
| ||
Granted |
|
|
|
|
|
| ||
Forfeited |
|
| ( | ) |
|
|
| |
Balances at December 31, 2020 |
|
|
|
|
|
| ||
Granted(1) |
|
|
|
|
|
| ||
Forfeited(2) |
|
| ( | ) |
|
|
| |
Exercised |
|
| ( | ) |
|
|
| |
Balances at September 30, 2021 |
|
|
|
|
|
| ||
Unvested options outstanding expected to vest (3) |
|
|
|
|
|
| ||
(1) |
(2) | Forfeited options represent unvested shares and vested, expired shares related to employee terminations. |
|
|
(3) | Estimated forfeitures only include known forfeitures to-date as the Company typically accounts for forfeitures as they occur due to a limited history of forfeitures. |
A summary of options outstanding as of September 30, 2021, is shown below:
Exercise Prices |
| Number of Shares Subject to Options Outstanding |
| Weighted-Average Remaining Contractual Term in Years |
| Number of Shares Subject to Options Fully Vested and Exercisable |
| Weighted-Average Remaining Contractual Term in Years |
$ |
| |
|
| |
| ||
$ |
| |
|
| |
| ||
$ |
| |
|
| |
| ||
$ |
| |
|
| |
| ||
|
| |
|
| |
|
| 17 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
Restricted stock unit activity under the Plan was as follows:
|
| Restricted Stock Units |
|
| Weighted-Average Grant Date Fair Value per Unit |
| ||
Unvested balance at January 1, 2020 |
|
| — |
|
| $ | — |
|
Granted |
|
|
|
|
|
| ||
Vested |
|
| ( | ) |
|
|
| |
Forfeited |
|
| ( | ) |
|
|
| |
Unvested balance at January 1, 2021 |
|
|
|
|
|
| ||
Granted |
|
|
|
|
|
| ||
Vested |
|
| ( | ) |
|
|
| |
Forfeited |
|
| ( | ) |
|
|
| |
Unvested Balance at September 30, 2021 |
|
|
|
|
|
| ||
During the three months ended September 30, 2021, and 2020, the Company recognized $
The Company recognizes as an expense the fair value of options granted to persons (currently consultants) who are neither employees nor non-employee directors. Stock-based compensation expense for consultants, recorded as research and development expense for the three months ended September 30, 2021, and 2020, was $
The fair value of options granted from inception to September 30, 2021, was based on the Black-Scholes option-pricing model assuming the following factors: 4.7 to 6.2 years expected term,
Stock option grants and fair values under the Plan was as follows:
|
| Three Months Ended September 30, |
|
| Nine Months Ended September 30, |
| ||||||||||
|
| 2021 |
|
| 2020 |
|
| 2021 |
|
| 2020 |
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Stock options granted |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Weighted-average grant date fair value per share |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
Fair value of shares vested |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
| 18 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
At September 30, 2021, the aggregate intrinsic value of outstanding vested stock options was approximately $
Note 5 - Related Party Transactions
As of September 30, 2021, Tactic Pharma, LLC (“Tactic Pharma”), the Company’s initial investor, beneficially owned
None of the related parties discussed in this paragraph received compensation other than market-rate salary, market-rate stock-based compensation and benefits and performance-based bonus or in the case of non-employee directors, market-rate Board fees and market-rate stock-based compensation. The Company considers the following individuals as related parties: Three of the Company’s board members were also Managing Members of Tactic Pharma as of September 30, 2021. Chandler D. Robinson is the Company’s Co-Founder, Chief Executive Officer, common stockholder, Managing Member of Tactic Pharma, former Manager of the predecessor LLC, Manager of CDR Pharma, LLC and Board member of Monopar as a C Corporation. Andrew P. Mazar is the Company’s Co-Founder, Executive Vice President of Research and Development, Chief Scientific Officer, common stockholder, Managing Member of Tactic Pharma, former Manager of the predecessor LLC and Board member of Monopar as a C Corporation. Michael Brown is a Managing Member of Tactic Pharma (as of February 1, 2019, with no voting power as it relates to Monopar), a previous managing member of Monopar as an LLC, common stockholder and Board member of Monopar as a C Corporation.
Note 6 – Commitments and Contingencies
License, Development and Collaboration Agreements
Onxeo S.A.
In June 2016, the Company executed an option and license agreement with Onxeo S.A. (“Onxeo”), a public French company, which gave Monopar the exclusive option to license (on a world-wide exclusive basis) Validive to pursue treating severe oral mucositis in patients undergoing chemoradiation treatment for head and neck cancers. The pre-negotiated Onxeo license agreement for Validive as part of
Under the agreement, the Company is required to pay royalties to Onxeo on a product-by-product and country-by-country basis until the later of (1) the date when a given product is no longer within the scope of a patent claim in the country of sale or manufacture, (2) the expiry of any extended exclusivity period in the relevant country (such as orphan drug exclusivity, pediatric exclusivity, new chemical entity exclusivity, or other exclusivity granted beyond the expiry of the relevant patent), or (3) a specific time period after the first commercial sale of the product in such country. In most countries, including the U.S., the patent term is generally 20 years from the earliest claimed filing date of a non-provisional patent application in the applicable country, not taking into consideration any potential patent term adjustment that may be filed in the future or any regulatory extensions that may be obtained. The royalty termination provision pursuant to (3) described above is shorter than 20 years and is the least likely cause of termination of royalty payments.
| 19 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
The Onxeo license agreement does not have a pre-determined term, but expires on a product-by-product and country-by-country basis; that is, the agreement expires with respect to a given product in a given country whenever the Company’s royalty payment obligations with respect to such product have expired. The agreement may also be terminated early for cause if either the Company or Onxeo materially breach the agreement, or if either the Company or Onxeo become insolvent. The Company may also choose to terminate the agreement, either in its entirety or as to a certain product and a certain country, by providing Onxeo with advance notice.
The Company is internally developing Validive and has activated clinical sites and begun patient dosing for its VOICE clinical trial, which, if successful, may allow the Company to apply for marketing approval within the next several years. The Company will need to raise significant funds or enter into a collaboration partnership to support the further development, including potential commercialization of Validive. As of September 30, 2021, the Company had not reached any of the pre-specified milestones and has not been required to pay Onxeo any funds under this license agreement other than the $1 million one-time license fee.
Grupo Español de Investigación en Sarcomas (“GEIS”)
In June 2019, the Company executed a clinical collaboration agreement with GEIS for the development of camsirubicin in patients with advanced soft tissue sarcoma (“ASTS”). Following completion of the Phase 1b clinical trial in the U.S. that Monopar initiated in the third quarter of 2021 with the first patient dosed in October 2021, the Company continues to expect that GEIS will sponsor and lead a multi-country, randomized, open-label Phase 2 clinical trial to evaluate camsirubicin head-to-head against doxorubicin, the current first-line treatment for ASTS.
XOMA Ltd.
The intellectual property rights contributed by Tactic Pharma to the Company included the non-exclusive license agreement with XOMA Ltd. for the humanization technology used in the development of MNPR-101. Pursuant to such license agreement, the Company is obligated to pay XOMA Ltd. clinical, regulatory and sales milestones for MNPR-101
| 20 |
| Table of Contents |
MONOPAR THERAPEUTICS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2021
Operating Leases
The Company is currently leasing office space for its executive headquarters at 1000 Skokie Blvd., in the Village of Wilmette, Illinois for $
During the three months ended September 30, 2021, and 2020, the Company recognized operating lease expenses of $
Legal Contingencies
The Company may be subject to claims and assessments from time to time in the ordinary course of business. No claims have been asserted to date.
Indemnification
In the normal course of business, the Company enters into contracts and agreements that contain a variety of representations and warranties and provide for general indemnification. The Company’s exposure under these agreements is unknown because it involves claims that may be made against the Company in the future, but that have not yet been made. To date, the Company has not paid any claims nor been required to defend any action related to its indemnification obligations. However, the Company may record charges in the future as a result of future claims against these indemnification obligations.
In accordance with its second amended and restated certificate of incorporation, amended and restated bylaws and the indemnification agreements entered into with each officer and non-employee director, the Company has indemnification obligations to its officers and non-employee directors for certain events or occurrences, subject to certain limits, while they are serving at the Company’s request in such capacities. There have been no indemnification claims to date.
| 21 |
| Table of Contents |
Item 2. Management's Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of our financial condition and results of operations together with our condensed consolidated financial statements and related notes contained in this Quarterly Report on Form 10-Q. Some of the information contained in this discussion and analysis are set forth elsewhere in this Quarterly Report on Form 10-Q, including information with respect to our plans and strategy for our business and related financing activities, includes forward-looking statements that involve risks and uncertainties.
Overview
We are a clinical stage biopharmaceutical company focused on developing proprietary therapeutics designed to extend life or improve quality of life for cancer patients. We are building a drug development pipeline through the licensing and acquisition of therapeutics in late preclinical and clinical development stages. We leverage our scientific and clinical experience to help reduce the risk of and accelerate the clinical development of our drug product candidates.
Validive Clinical Update
In February 2021, we dosed the first patient in our Phase 2b/3 VOICE trial of Validive® for the prevention of chemoradiation treatment (“CRT”)-induced severe oral mucositis in patients with oropharyngeal cancer (“VOICE”). In August 2021, we successfully reached our target of 20 activated clinical trial sites for the Phase 2b portion of the 2b/3 Validive® VOICE trial and in September 2021, we received authorization to proceed with the VOICE clinical trial in multiple countries in Europe. We plan to continue to activate additional sites in both the US and the EU.
Given the COVID-19 pandemic and its effects on clinical trials, we have adjusted our clinical development plans accordingly to fit what is feasible in the current environment. We have simplified the design of the previously planned Phase 3 clinical trial for our lead product candidate, Validive, to a seamless, adaptive Phase 2b/3 clinical trial design (our VOICE trial) that will allow us to minimize touch points with patients and sites. This trial design will allow us to immediately advance to the Phase 3 portion of the trial if supported by the interim data at the end of the Phase 2b portion of the trial. To complete the VOICE clinical program, including, if required, completing a second Phase 3 confirmatory clinical trial, we will require additional funding in the millions or tens of millions of dollars (depending on if we have consummated a collaboration or partnership or neither for Validive), which we are planning to pursue within the next 12 months.
Camsirubicin Clinical Update
In August 2021, we announced clearance from the U.S. Food and Drug Administration (“FDA”) to proceed with an open-label Phase 1b dose-escalation clinical trial evaluating camsirubicin plus growth factor support (pegfilgrastim/G-CSF) in patients with advanced soft tissue sarcoma (“ASTS”). In September 2021 we initiated the Phase 1b clinical trial, and in October 2021 we dosed the first patients. Following completion of the Phase 1b clinical trial, we continue to expect that Grupo Español de Investigación en Sarcomas (“GEIS”) will sponsor and lead a multi-country, randomized, open-label Phase 2 clinical trial to evaluate camsirubicin head-to-head against doxorubicin, the current first-line treatment for ASTS. We believe we have funds sufficient to obtain topline results from the Phase 1b clinical trial. Additional funding will be required to support further development beyond our Phase 1b clinical trial.
Financial Status
Under our Capital on DemandTM Sales Agreement with JonesTrading Institutional Services, LLC (“JonesTrading”), through September 30, 2021, we sold 1,964,724 shares of our common stock at an average gross price of $10.02 per share for net proceeds of $19,100,602, after fees and commissions of $591,188. We did not sell any shares of our common stock under this agreement during the three months ended September 30, 2021. The maximum aggregate offering price under the agreement has been reached and we do not expect further sales under this agreement. The balance of cash and cash equivalents as of September 30, 2021, was $22,341,564.
MNPR-101 RIT Development Update
Pursuant to our 50/50 collaboration development agreement with NorthStar Medical Radioisotopes, LLC (“NorthStar”) to develop potential radioimmunotherapeutics (“RITs”) to treat severe COVID-19 (patients with SARS-CoV-2 infection), we have coupled MNPR-101 to therapeutic radioisotopes supplied by NorthStar. The resulting conjugates are designed to be highly selective agents that have the potential to kill aberrantly activated cytokine-producing immune cells. By eradicating these cells with a uPAR-targeted RIT (“uPRIT”), the therapeutic goal is to spare healthy cells while quickly reducing the cytokine storm and its harmful systemic effects. Through October 31, 2021, we have filed several patent applications on discoveries made through the NorthStar collaboration, and have incurred immaterial expense related to the collaboration, while partnering with several key companies and institutions to further the collaboration’s development efforts. These collaborators include: IsoTherapeutics Group, LLC, which generated the uPRIT candidates; Aragen Bioscience Inc., which screened the uPRIT candidates through preclinical biochemical testing; and Texas Lung Injury Institute / University of Texas Health Science Center at Tyler, which plans to perform preclinical testing and, if successful, clinical testing. In addition, Monopar and NorthStar have advanced their collaboration to investigate MNPR-101 coupled to diagnostic radioisotopes as a companion diagnostic for uPRIT for use in COVID-19 and advanced cancers.
| 22 |
| Table of Contents |
In February 2021, we announced the publication of a peer-reviewed study in the European Journal of Cancer which reported the potential utility of MNPR-101 conjugates as uPAR imaging agents to improve surgical outcomes in bladder cancer and for surveillance post-resection. This publication builds on previous studies using conjugates of MNPR-101 and its mouse analog, ATN-658, for the optical imaging of oral and colon cancer.
In March 2021, we announced the publication of a peer-reviewed study titled “Engineered Antibody Fragment against the Urokinase Plasminogen Activator for Fast Delineation of Triple-Negative Breast Cancer by Positron Emission Tomography.” Urokinase plasminogen activator (“uPA”) is an established biomarker in current breast cancer clinical practice guidelines and its presence is used to select appropriate drug treatment. This study demonstrates the potential to identify breast cancers with uPA overexpression and monitor uPA activity during treatment using positron emission tomography or PET imaging along with our uPA antibody fragment radiotracer. We have a panel of proprietary antibodies and antibody fragments to uPA and its receptor uPAR (such as MNPR-101).
MNPR-202 and Related Analogs Updates
In June 2021, we entered into a collaboration agreement with the Cancer Science Institute of Singapore (“CSI Singapore”), one of Asia’s premier cancer research centers, at the National University of Singapore (“NUS”) (consistently ranked as one of the world's top universities) to evaluate the activity of MNPR-202 and related analogs in multiple types of cancer. MNPR-202 was designed to retain the same potentially non-cardiotoxic backbone as camsirubicin but is modified at other positions which may enable it to work in certain cancers that are resistant to camsirubicin and doxorubicin. In December 2020, we announced the issuance of our composition of matter U.S. patent (US10,450,340) covering MNPR-202 and related analogs. CSI Singapore will explore the mechanism of action of MNPR-202 and related analogs in preclinical cancer models in order to guide the rational design of MNPR-202 drug combinations including immunotherapy-MNPR-202 for the treatment of cancer.
Patent Updates
In May 2021, we and NorthStar filed a provisional patent application with the U.S. Patent and Trademark Office (“USPTO”) titled “Bio-Targeted Radiopharmaceutical Compositions Containing Ac-225 and Methods of Preparation.” Radiopharmaceutical therapy is a promising approach to treat cancer and other diseases using radioactive metals bound with proteins/antibodies to target and kill cells. Actinium-225 (“Ac-225”) is emerging as a radioactive isotope of choice for radiopharmaceuticals due to favorable properties such as its long half-life, high potency, and induction of localized cell death. This provisional patent relates to the unexpected observation by us and NorthStar that using the metal binding agent 3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-1(15),11,13-triene-3,6,9-triacetic acid (“PCTA”) to attach Ac-225 to MNPR-101 resulted in nearly 100% binding of Ac-225 to the PCTA-MNPR-101 conjugates. If validated through further evaluation, it could potentially improve efficacy and safety and enhance manufacturing efficiency of Actinium-based radiopharmaceuticals, the full potential of which are presently constrained by the price and scarcity of Ac-225.
Also in May 2021, we and NorthStar filed a provisional composition of matter patent application titled “Urokinase Plasminogen Activator Receptor-Targeted Radiopharmaceutical” covering a radiotherapeutic consisting of our proprietary antibody MNPR-101 bound to Ac-225 via the metal binding agent PCTA. This RIT demonstrated 98% radiochemical purity and high stability and has the potential to be a highly selective, potent treatment for a variety of cancers, severe COVID-19, and other diseases characterized by aberrant uPA receptor expression.
| 23 |
| Table of Contents |
Our Product Pipeline
Our Product Candidates
Validive (clonidine hydrochloride mucobuccal tablet; clonidine HCI MBT)
Validive is a mucobuccal tablet (“MBT”) formulation of clonidine. The MBT formulation was developed to enhance oral mucosal drug delivery and provide high salivary concentrations of the active ingredient while minimizing systemic absorption. The Validive tablet is tasteless and self-administered once daily by affixing it to the outside of the upper gum where it dissolves slowly over the period of several hours, resulting in the extended release of clonidine into the oral cavity and oropharynx, the site of severe oral mucositis (“SOM”) following chemoradiation treatment (“CRT”) for oropharyngeal cancer (“OPC”). Validive therapy is designed to begin on the first day of CRT and continue daily through the last day of CRT.
SOM is the painful and debilitating inflammation and ulceration of the mucous membranes lining the oral cavity and oropharynx in response to chemoradiation therapy. The majority of patients receiving CRT to treat their OPC develop SOM, which is one of the most common and devastating side effects of treatment in this indication. We believe Validive has the potential to address several critical elements that affect SOM patients, including:
Reduction in the incidence of SOM. SOM can increase the risk of acute and chronic comorbidities, including dysphagia, trismus and lung complications, which are often irreversible and lead to increased hospitalization and the need for additional interventions. In a Phase 2 clinical trial, the OPC patient cohort treated with Validive 100 µg demonstrated a reduction in the absolute incidence of SOM compared to placebo of 26.3% (incidence rate of 65.2% in placebo, 45.0% in Validive 50 µg group, 38.9% in Validive 100 µg group). A reduced incidence of SOM in OPC patients may lower the risk of acute and chronic comorbidities, improve clinical outcomes and quality of life.
Delay in the time to onset of SOM. SOM can cause cancer treatment delay and/or discontinuation, which may impact overall survival outcomes. In the Phase 2 clinical trial, OPC patients had a time to onset of SOM of 37 days in the placebo cohort; 45-day time to onset of SOM in the Validive 50 µg cohort; and median was not reached in the Validive 100 µg group as fewer than half of the patients developed SOM. Prolonging time to onset of SOM may lead to fewer missed CRT treatments, resulting in improved overall survival outcomes.
Decrease in the duration of SOM. Longer duration of SOM leads to a higher risk of the need for parenteral nutrition and lower quality of life. SOM patients experience difficulty or inability to drink and/or eat, and difficulty in swallowing often results in malnourishment and feeding tube intervention. The Phase 2 clinical trial data demonstrated a 15.5-day reduction (by 37.8%) in the duration of SOM for patients treated with Validive 100 µg (41-day median duration with placebo, 34 days with the Validive 50 µg group, and 25.5 days for the Validive 100 µg group) in patients that developed SOM. Median duration across all patients, inclusive of both those that did and did not develop SOM, was 17 days in the placebo group and 0 days in each of the Validive 50 ug and 100 µg groups. Reduced duration of SOM may result in lower risk of malnourishment and feeding tube intervention, and fewer treatment terminations/delays.
| 24 |
| Table of Contents |
In September 2017, we exercised an option to license Validive from Onxeo S.A., the company that had developed Validive through its Phase 2 clinical trial. In this completed Phase 2 clinical trial, Validive demonstrated clinically meaningful and dose-dependent efficacy signals within the 64-patient OPC population randomized to placebo. Additionally, patients in the Validive cohorts in the Phase 2 clinical trial demonstrated a safety profile similar to that of placebo. While not designed by us, Onxeo’s promising preclinical studies and Phase 2 clinical trial have informed the design and conduct of what we believe will be an effective Phase 2b/3 (VOICE) clinical trial.
SOM typically arises in the immune tissue at the back of the tongue and throat, which comprise the oropharynx, and consists of acute severe tissue damage and pain that prevents patients from swallowing, eating and drinking. Validive stimulates the alpha-2 adrenergic receptor (alpha-2AR) on macrophages (white blood cells present in the immune tissues of the oropharynx) suppressing pro-inflammatory cytokine expression. Validive exerts its effects locally in the oral cavity and oropharynx over a prolonged period of time through its unique MBT formulation. Patients who develop SOM are also at increased risk of developing late-onset toxicities, including trismus (jaw, neck, and throat spasms), dysphagia (disruption of a patient’s ability to eat and drink due to difficulty in swallowing), and lung complications, which are often irreversible and lead to increased hospitalization and the need for further interventions sometimes years after completion of CRT. We believe that a reduction in the incidence and duration of SOM by Validive will have the potential to reduce treatment discontinuation and/or treatment delays potentially leading to improved survival outcomes, and reducing or eliminating long-term morbidities resulting from CRT.
The OPC target population for Validive is the most rapidly growing segment of head and neck cancer (“HNC”) patients, estimated to be above 40,000 new cases annually of OPC in the U.S alone. The growth in OPC is driven by the increasing prevalence of oral human papilloma virus (“HPV”) infections in the U.S. and around the world. Despite the availability of a pediatric/adolescent HPV vaccine, the rate of OPC incidence in adults is not anticipated to be materially reduced for decades due to low adoption of the vaccine to date. As a result, the incidence of HPV-driven OPC is projected to increase for many years to come and will continue to support a clinical need for Validive for the prevention of CRT-induced SOM in patients with OPC since CRT is the standard of care treatment, and we do not anticipate this changing for years to come.
A pre-Phase 3 meeting with the FDA was held, and based on the meeting discussion a Phase 3 clinical protocol and accompanying statistical analysis plan (“SAP”) was submitted to the FDA for review and comments. We have also received protocol assistance and advice on our Phase 3 protocol and SAP from the European Medicines Agency Committee on Human Medicinal Products (EMA/CHMP/SAWP). Based on comments and guidance provided by FDA and EMA, and our analysis of the current COVID-19 pandemic and its effects on clinical trials, we have modified our original adaptive design Phase 3 clinical trial to be a seamless Phase 2b/3 (VOICE) clinical trial to better fit the current clinical research environment. The primary endpoint, absolute incidence of SOM, remains the same, but the overall design of the trial has been simplified and the touch points with the healthcare system have been minimized. We have now initiated clinical trial sites and commenced dosing in the Phase 2b portion of our VOICE trial. We anticipate the interim completion of Phase 2b portion of the VOICE trial will be reached in the first half of 2022, and the Phase 3 enrollment will be completed in the first half of 2023. We will need to raise additional funding or find a suitable pharmaceutical partner to complete the VOICE clinical program including, if required, completion of a second Phase 3 confirmatory clinical trial. Validive has been granted fast track designation in the U.S., orphan drug designation in the EU, and has global intellectual property patent protection through mid-2029 not accounting for possible extensions.
Camsirubicin (5-imino-13-deoxydoxorubicin; formerly MNPR-201, GPX-150)
Camsirubicin is a proprietary doxorubicin analog. Doxorubicin is widely used to treat adult and pediatric solid and blood (hematologic) cancers, including soft tissue sarcomas, breast, gastric, ovarian and bladder cancers, leukemias and lymphomas. Despite clinical studies demonstrating the anti-cancer benefit of higher cumulative doses of doxorubicin, the clinical efficacy of doxorubicin has historically been limited by the risk of patients developing irreversible, potentially life-threatening cardiotoxicity at higher cumulative doses of drug. For example, several clinical studies completed in the 1990s demonstrated that concurrent doxorubicin (60 mg/m2, 8 cycles) and paclitaxel gave a 94% overall response rate in patients with metastatic breast cancer but led to 18% of these patients developing congestive heart failure. Reduction of doxorubicin to 4-6 cycles of treatment decreased the incidence of congestive heart failure, but also reduced response rates to 45-55%. In a clinical study looking at dose response, sarcoma patients on the high dose (75 mg/m2) doxorubicin had a response rate of 37% compared to just 18% in the low dose (45 mg/m2) doxorubicin group. With the cumulative dose restriction on doxorubicin, the median progression free survival for ASTS patients is approximately 6 months, with median overall survival of 12-15 months. There is a significant unmet opportunity to develop a replacement for doxorubicin that can be dosed higher and for longer.
Camsirubicin has been engineered specifically to retain the anticancer activity of doxorubicin while minimizing the toxic effects on the heart. Similar to doxorubicin, the antitumor effects of camsirubicin are mediated through the stabilization of the topoisomerase II complex after a DNA strand break and DNA intercalation leading to tumor cell apoptosis (cell death). Inhibiting the topoisomerase II-alpha isoform is desired for the anti-cancer effect, while inhibiting the topoisomerase II-beta isoform has been demonstrated to mediate, at least in part, the cardiotoxicity associated with doxorubicin. Camsirubicin is more selective than doxorubicin for inhibiting topoisomerase II-alpha versus topoisomerase II-beta. This selectivity may at least partly explain the minimal cardiotoxicity that has been observed for camsirubicin in preclinical and clinical studies to date. We believe these attributes provide a strong rationale to develop camsirubicin without restriction on cumulative dose, in a broad spectrum of cancer types.
| 25 |
| Table of Contents |
A Phase 2 clinical trial for camsirubicin has been completed in patients with ASTS. In this study, 52.6% of patients evaluable for tumor progression demonstrated clinical benefit (partial response or stable disease), which was proportional to dose and consistently observed at higher cumulative doses of camsirubicin (>1000 mg/m2). Camsirubicin was very well tolerated in this study and underscored the ability to potentially administer camsirubicin without restriction of cumulative dose in patients with ASTS. Although doxorubicin has been the standard of care treatment for over 40 years for patients with ASTS, doxorubicin is limited to a lifetime cumulative dose maximum of 450 mg/m2. This means that even if a patient is responding, they are pulled off of doxorubicin treatment once this cumulative dose has been reached. Thus, there is a significant unmet opportunity to develop a replacement for doxorubicin that retains anti-cancer activity while reducing or eliminating the risk for irreversible heart damage.
Based on encouraging clinical results to date, we plan to continue the development of camsirubicin as first-line treatment in patients with ASTS, where the current first-line treatment is doxorubicin. The aim is to administer camsirubicin without restricting cumulative dose, thereby potentially improving efficacy beyond that of doxorubicin by continuing to treat patients who are responding to treatment.
In June 2019, we executed a clinical collaboration agreement with GEIS for the development of camsirubicin in patients with ASTS. GEIS is an internationally renowned non-profit organization focused on the research, development and management of clinical trials for sarcoma that has worked with many of the leading biotech and global pharmaceutical companies. Based on our inability to gain timely regulatory approval to initiate the planned camsirubicin dose-escalation run-in and Phase 2 clinical trial in Spain, we submitted an IND with the FDA and gained clearance to conduct a Phase 1b dose escalation clinical trial in ASTS patients in the U.S. In September 2021 we initiated the Phase 1b clinical trial in the U.S., and in October 2021 we dosed the first patient. Following completion of the Phase 1b clinical trial, we continue to expect that GEIS will sponsor and lead a multi-country, randomized, open-label Phase 2 clinical trial to evaluate camsirubicin head-to-head against doxorubicin, the current first-line treatment for ASTS. We believe we have funds sufficient to obtain topline results from the Phase 1b clinical trial. Additional funding will be required to support further development beyond the Phase 1b clinical trial. Camsirubicin has been granted orphan drug designation for the treatment of soft tissue sarcoma in the U.S. and the EU.
MNPR-101 (formerly huATN-658)
MNPR-101 is a novel, preclinical stage drug candidate. It is a first-in-class humanized monoclonal antibody to the urokinase plasminogen activator receptor (“uPAR”), a well-credentialed cancer therapeutic target. uPAR is a protein receptor that resides on the cell surface and is overexpressed in many deadly cancers, but has little to no expression in healthy tissue; several Phase 1 imaging studies utilizing a peptide against uPAR in advanced cancer patients show that uPAR is detected selectively in the tumor.
In normal cells, uPAR is transiently expressed as part of a highly regulated process required for the breakdown of the extracellular matrix during normal tissue remodeling. In cancer, however, uPAR is constitutively overexpressed by the tumor cell, and the uPAR extracellular matrix degrading function is hijacked by the tumor to support tissue invasion, metastasis, and angiogenesis. uPAR expression is important to tumor cell survival, and uPAR expression increases in high grade and metastatic disease.
MNPR-101 has demonstrated significant antitumor activity in numerous preclinical models of tumor growth, both as a monotherapy and in combination with other therapeutics. Based on the selective expression of uPAR in numerous tumor types, we anticipate MNPR-101 will be well-tolerated and amenable to a variety of combination treatment approaches in the clinic.
| 26 |
| Table of Contents |
uPRIT as a Potential Therapeutic for Severe COVID-19
A radioimmunotherapeutic (“RIT”) based on MNPR-101 is being developed for the treatment of severe COVID-19 and cancer. We have entered into a collaboration development agreement with NorthStar to develop potential uPRITs to treat severe COVID-19. This collaboration combines NorthStar’s expertise in the innovative production, supply, and distribution of important medical radioisotopes with our expertise in therapeutic drug development. We have coupled MNPR-101 with a therapeutic radioisotope in collaboration with NorthStar and IsoTherapeutics Group, LLC, which have generated the MNPR-101 RIT conjugates. uPAR seems to be selectively expressed on aberrantly activated immune cells. In response to coronavirus (SARS-CoV2) infection, these rogue immune cells produce pro-inflammatory cytokines that can cause runaway inflammation throughout the body, commonly referred to as a “cytokine storm.” It is this systemic hyper-inflammatory state that is thought to be largely responsible for the severe lung injury and further multiple organ damage that contributes to poor outcomes and death in patients with severe COVID-19.
In collaboration with NorthStar, we filed a provisional patent application entitled “Precision Radioimmunotherapeutic Targeting of the Urokinase Plasminogen Activator Receptor (uPAR) for Treatment of Severe COVID-19 Disease” with the USPTO on June 15, 2020. A full international application (International Application Number PCT/US2021/037416) that claims priority to the provisional filing date was filed under the Patent Cooperation Treaty (“PCT”) on June 15, 2021. This application covers novel compositions and uses of cytotoxic radioisotopes attached to antibodies that bind to uPAR, thereby creating precision targeted radiotherapeutics, also known as uPRITs, for the treatment of severe COVID-19 and other respiratory diseases. Advanced COVID-19 patients frequently develop severe, life-threatening, pulmonary inflammation as a result of a viral induced cytokine storm. The development of this cytokine storm is associated with a high rate of mortality in severe COVID-19 patients, even when oxygen support and mechanical ventilation are utilized. uPRITs have been designed with the goal of selectively eradicating the aberrantly activated immune cells responsible for causing cytokine storm and its harmful systemic effects. The co-inventors of the provisional patent application are James Harvey, Chief Scientific Officer of NorthStar, and Andrew P. Mazar, our Chief Scientific Officer.
In May 2021, we and NorthStar filed a provisional patent application with the USPTO titled “Bio-Targeted Radiopharmaceutical Compositions Containing Ac-225 and Methods of Preparation.” Radiopharmaceutical therapy is a promising approach to treat cancer and other diseases using radioactive metals bound with proteins/antibodies to target and kill cells. If validated through further evaluation, it could potentially improve efficacy and safety and enhance manufacturing efficiency of Actinium-based radiopharmaceuticals, the full potential of which are presently constrained by the price and scarcity of Ac-225.
Also, the provisional composition of matter patent application titled “Urokinase Plasminogen Activator Receptor-Targeted Radiopharmaceutical” covering a radiotherapeutic consisting of our proprietary antibody MNPR-101 bound to Ac-225 via the metal binding agent PCTA has the potential to be a highly selective, potent treatment for a variety of cancers, severe COVID-19, and other diseases characterized by aberrant uPAR expression.
In addition to NorthStar and the IsoTherapeutics Group, LLC, which generated the uPRIT candidates, we have entered into collaborations with: Aragen Bioscience Inc., which screened the uPRIT candidates through preclinical biochemical testing; and Texas Lung Injury Institute / University of Texas Health Science Center at Tyler, which plans to perform preclinical testing and, if successful, clinical testing.
uPRIT as a Potential Therapeutic for Cancer
Monopar is also developing uPRITs, cytotoxic radioisotopes attached to antibodies that bind to uPAR, as precision targeted radioimmunotherapeutics for cancer. uPAR is a protein receptor that resides on the cell surface and is expressed in many deadly cancers, but has little to no expression in healthy tissue. Monopar is conducting preclinical development of uPRITs and companion diagnostics, which consist of imaging radioisotopes attached to antibodies that bind to uPAR, for the potential diagnosis and treatment of advanced cancers.
MNPR-101 as a Potential Imaging Agent
Using MNPR-101, a multimodal imaging probe was developed and tested in vivo in human bladder cancer models. Bladder cancer is often treated with transurethral resection to remove cancerous tissue; however, recurrence can occur in up to 78% of patients within 5 years. Up to 40% of recurrent cases develop muscle invasive disease, which has a poor prognosis and requires complete removal of the bladder. Many patients with muscle-invasive bladder cancer go on to develop and succumb to metastatic disease. A publication in the European Journal of Cancer (Baart et al. 2021) reported that high expression of uPAR in bladder cancer is localized at the tumor periphery, suggesting that using a fluorescent-conjugated MNPR-101 probe might allow surgeons to better visualize the borders of the tumor, potentially resulting in more complete tumor resection and thereby minimizing relapse. Similar approaches have been utilized successfully in the resection of other tumor types, such as breast cancer.
| 27 |
| Table of Contents |
uPA and its receptor uPAR work together to drive aggressive tumor invasion, leading to metastasis, morbidity, and mortality in breast and other cancers. However, the amount of uPA in tumor is difficult to measure and currently requires a substantial amount of fresh frozen tissue. In a recent peer-reviewed publication, our antibody fragment (ATN-291 F(ab’)2) conjugated to a copper radiotracer enabled rapid PET visualization of tumors with uPA overexpression in a human breast cancer model in mice. PET imaging may expand the current application of uPA as a breast cancer biomarker non-invasively without the need to obtain substantial amounts of tumor biopsy tissue and enable the monitoring of tumor uPA levels during treatment. The publication demonstrates, and we may further research, the potential utility of our uPA antibody fragments as imaging agents in breast and other cancers.
MNPR-202
MNPR-202 is a camsirubicin analog, designed to retain the same potentially non-cardiotoxic backbone as camsirubicin but is modified at other positions which may enable it to be efficacious in certain cancers that are resistant to camsirubicin and doxorubicin. MNPR-202 and related analogs are covered under a newly issued U.S. composition of matter patent (US10,450,340).
In collaboration with CSI Singapore, we plan to evaluate the activity of MNPR-202 and related analogs in preclinical models of cancer.
Our Strategy
Our management team has extensive experience in developing therapeutics and medical technologies through global regulatory approval and commercialization. In aggregate, companies they co-founded have achieved four drug approvals and three diagnostic medical imaging device approvals in the U.S. and the EU, successfully sold an asset developed by management which is currently in Phase 3 clinical trials, sold two oncology-focused diagnostic imaging businesses to Fortune Global 1000 firms, and completed the clinical and commercial development and ultimately the sale of a commercial biopharmaceutical company for over $800 million in cash. In addition, the team has supported multiple regulatory submissions with the FDA and EMA and launched multiple drugs in the U.S and the EU. Understanding the preclinical, clinical, regulatory and commercial development processes and hurdles are key factors in successful drug development and the expertise demonstrated by our management team across all of these areas increases the probability of success in advancing the product candidates in our product pipeline. Our strategic goal is to acquire, develop and commercialize promising oncology product candidates that address important unmet medical needs of cancer patients. Seven key elements of our strategy to achieve this goal are to:
· | Leverage data generated from the Phase 2 Validive clinical trial to execute a successful VOICE clinical program for Validive for SOM in OPC. In the Phase 2 clinical trial the absolute incidence of SOM in OPC patients was reduced by 26.3%, the time to SOM onset was delayed, and the duration of disease in patients that developed SOM was decreased by 15.5 days in the Validive 100 µg cohort versus placebo. In addition to the data from the Phase 2 clinical trial, we believe the guidance from our key opinion leaders (“KOLs”) as well as from the FDA and EMA, and our own internal clinical trial design expertise, position us well for an effective VOICE clinical trial program. |
|
|
· | Obtain FDA and EMA approval of Validive to maximize the commercial potential of Validive in both the U.S. and the EU, and seek partnerships outside these markets. If the VOICE clinical program of Validive is successful and FDA and EMA approvals are obtained, we currently intend to commercialize Validive in the U.S. and the EU ourselves, which may include establishing our own specialty sales force and seeking partnerships outside of these territories for regulatory approval and drug sales and distribution. |
|
|
· | Advance the clinical development of camsirubicin, by pursuing indications where doxorubicin has demonstrated efficacy. ASTS will be the first indication, which will allow camsirubicin to go head-to-head against doxorubicin, the current first-line treatment. In this indication, camsirubicin previously demonstrated clinical benefit (stable disease or partial response) in 52.6% of patients evaluable for tumor progression in a single-arm Phase 2 study. Clinical benefit was proportional to dose and was consistently observed at higher cumulative doses of camsirubicin (>1000 mg/m2). Camsirubicin was very well tolerated in this Phase 2 study and underscored the ability to potentially administer camsirubicin without restriction as to cumulative dose (doxorubicin is limited due to heart toxicity to 450 mg/m2 cumulative dose). |
|
|
· | Continue the development of MNPR-101, MNPR-101 RIT and related molecules as therapeutic, diagnostic and imaging agents. We plan to continue the development of MNPR-101, MNPR-101 RIT and related molecules for diagnostic, imaging, and therapeutic use in severe COVID-19 and in cancer. |
|
|
· | Continue the development of MNPR-202 and related analogs in multiple types of cancers. The 2-pyrrilino camsirubicin analog (MNPR-202) and related analogs represent proprietary compositions of matter designed to retain the non-cardiotoxic backbone of camsirubicin yet exhibit novel features in terms of antitumor activity and mechanism that distinguish these analogs from camsirubicin as well as from doxorubicin. |
| 28 |
| Table of Contents |
· | Expand our drug development pipeline through advancing current assets, in-licensing, and acquisition of oncology product candidates. We plan to continue the expansion of our drug development pipeline through acquiring or in-licensing additional oncology product candidates, particularly those that leverage existing scientific and clinical data that helps reduce the risks of the next steps in clinical development. |
|
|
· | Utilize the expertise and prior experience of our team in the areas of asset acquisition, drug development and commercialization to establish ourselves as a leading biopharmaceutical company. Our senior executive team has relevant experience in biopharmaceutical in-licensing and acquisitions as well as developing product candidates through approval and commercialization. In aggregate, our team has co-founded BioMarin Pharmaceutical (Nasdaq: BMRN), Sensant Corp (acquired by Siemens), American BioOptics (assets acquired by Olympus), Raptor Pharmaceuticals ($800 million sale to Horizon Pharma), and Tactic Pharma, LLC (“Tactic Pharma”) (sale of lead asset, choline tetrathiomolybdate, which was ultimately acquired by Alexion in June 2018 for $764 million; Alexion was recently acquired by AstraZeneca. |
Revenues
We are an emerging growth company. We have no approved drugs and have not generated any revenues. To date, we have engaged in acquiring or in-licensing pharmaceutical drug product candidates, entering into collaboration agreements for testing and clinical development of our drug product candidates and providing the infrastructure to support the clinical development of our drug product candidates. We do not anticipate commercial revenues from operations until we complete testing and development of one of our drug product candidates and obtain marketing approval or we sell, enter into a collaborative marketing arrangement, or out-license one of our drug product candidates to another party. See “Liquidity and Capital Resources”.
Recently Issued and Adopted Accounting Pronouncements
During the three months ended September 30, 2021, there were no relevant recently issued accounting pronouncements that would impact our financial position and our condensed consolidated results of operations and comprehensive loss.
Critical Accounting Policies and Use of Estimates
While our significant accounting policies are described in more detail in Note 2 of our condensed consolidated financial statements included elsewhere in this Quarterly Report on Form 10-Q, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our condensed consolidated financial statements.
Clinical Trials Expense
We accrue and expense the costs for clinical trial activities performed by third parties based upon estimates of the percentage of work completed over the life of the individual study in accordance with agreements established with contract research organizations, service providers, and clinical trial sites. We estimate the amounts to accrue based upon discussions with internal clinical personnel and external service providers as to progress or stage of completion of trials or services and the agreed upon fee to be paid for such services. Costs of setting up clinical trial sites for participation in the trials are expensed immediately as R&D expenses. Clinical trial site costs related to patient screening and enrollment are accrued as patients are screened/entered into the trial.
Stock-Based Compensation
We account for stock-based compensation arrangements with employees, non-employee directors and consultants using a fair value method, which requires the recognition of compensation expense for costs related to all stock-based awards, including stock option grants and restricted stock units (“RSUs”). The fair value method requires us to estimate the fair value of stock-based payment awards on the date of grant using an option pricing model or the closing stock price on the date of grant in the case of RSUs.
| 29 |
| Table of Contents |
Stock-based compensation costs for stock awards granted to our employees, non-employee directors and consultants are based on the fair value of the underlying instruments calculated using the Black-Scholes option-pricing model on the date of grant for stock options and using the closing stock price on the date of grant for RSUs and recognized as expense on a straight-line basis over the requisite service period, which is the vesting period. Determining the appropriate fair value model and related assumptions requires judgment, including selecting methods for estimating the Company’s future stock price volatility, forfeiture rates and expected term. The expected volatility rates are estimated based on the actual volatility of comparable public companies over recent historical periods of the same length as the expected term. We generally selected these companies based on reasonably comparable characteristics, including market capitalization, risk profiles, stage of corporate development and with historical share price information sufficient to meet the expected term of the stock-based awards. The expected term for stock options granted during the three and nine months ended September 30, 2021, and 2020 was estimated using the simplified method. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We have not paid dividends and do not anticipate paying a cash dividend in future vesting periods and, accordingly, use an expected dividend yield of zero. The risk-free interest rate is based on the rate of U.S. Treasury securities with maturities consistent with the estimated expected term of the awards.
Results of Operations
Comparison of the Three and Nine Months Ended September 30, 2021, and 2020
The following table summarizes the results of our operations for the three and nine months ended September 30, 2021, and 2020:
|
| Three Months Ended September 30, |
|
| Nine Months Ended September 30, |
| ||||||||||||||||||
|
| (Unaudited) |
|
| (Unaudited) |
| ||||||||||||||||||
(in thousands) |
| 2021 |
|
| 2020 |
|
| Variance |
|
| 2021 |
|
| 2020 |
|
| Variance |
| ||||||
Research and development expenses |
| $ | 1,827 |
|
| $ | 1,256 |
|
| $ | 571 |
|
| $ | 4,511 |
|
| $ | 2,433 |
|
| $ | 2,078 |
|
General and administrative expenses |
|
| 632 |
|
|
| 392 |
|
|
| 240 |
|
|
| 1,935 |
|
|
| 1,815 |
|
|
| 120 |
|
Total operating expenses |
|
| 2,459 |
|
|
| 1,648 |
|
|
| 811 |
|
|
| 6,446 |
|
|
| 4,248 |
|
|
| 2,198 |
|
Operating loss |
|
| (2,459 | ) |
|
| (1,648 | ) |
|
| (811 | ) |
|
| (6,446 | ) |
|
| (4,248 | ) |
|
| (2,198 | ) |
Interest income |
|
| 1 |
|
|
| 9 |
|
|
| (8 | ) |
|
| 23 |
|
|
| 72 |
|
|
| (49 | ) |
Net loss |
| $ | (2,458 | ) |
| $ | (1,639 | ) |
| $ | (819 | ) |
| $ | (6,423 | ) |
| $ | (4,176 | ) |
| $ | (2,247 | ) |
Research and Development Expenses
Research and Development (“R&D”) expenses for the three months ended September 30, 2021, were $1,827,000, compared to $1,256,000 for the three months ended September 30, 2020. This represents an increase of $571,000 primarily attributed to increases in expenses of $452,000 for Validive clinical trial expenses and $210,000 due to annual R&D personnel salary increases, annual (non-cash) equity grants, salaries and benefits of new R&D personnel net of a decrease in CEO's salary and benefits allocated to R&D expenses and $18,000 in net increases to other R&D expenses offset by a decrease of $109,000 in camsirubicin regulatory and manufacturing-related costs.
R&D expenses for the nine months ended September 30, 2021, were $4,511,000, compared to $2,433,000 for the nine months ended September 30, 2020. This represents an increase of $2,078,000 primarily attributed to increases in expenses of $975,000 for Validive clinical trial expenses, $723,000 due to annual R&D personnel salary increases, annual (non-cash) equity grants, salaries and benefits of new R&D personnel and an increase in the CEO's salary and benefits allocated to R&D expenses due to significant efforts related to the start-up of clinical trial activities, $303,000 for the planning and initiation of the camsirubicin Phase 1b clinical trial including regulatory and manufacturing-related costs, and $77,000 in net increases to other R&D expenses.
General and Administrative Expenses
General and administrative (“G&A”) expenses for the three months ended September 30, 2021, were $632,000, compared to $392,000 for the three months ended September 30, 2020. This represents an increase of $240,000 primarily attributable to increases in expenses of $201,000 for annual G&A personnel salary increases, annual (non-cash) equity grants and an increase of the CEO's salary and benefits allocated to G&A and $43,000 in patent expenses offset by $4,000 in net decreases to other G&A expenses.
G&A expenses for the nine months ended September 30, 2021, were $1,935,000, compared to $1,815,000 for the nine months ended September 30, 2020. This represents an increase of $120,000 primarily attributed to increases in expenses of $96,000 for patent expenses, $66,000 for annual G&A personnel salary increases, annual (non-cash) equity grants offset by a decrease in the CEO's salary and benefits allocated to G&A, $36,000 for G&A consulting and $7,000 for net increases to other G&A expenses offset by $85,000 for the reduction in stock-based compensation for non-employee directors.
| 30 |
| Table of Contents |
Interest Income
Interest income for the three months ended September 30, 2021, decreased by $8,000 versus the three months ended September 30, 2020, due to a significant decrease in bank interest rates offset by an increase in bank balances resulting from funds raised in our Capital on DemandTM Sales Agreement with JonesTrading during the first quarter of 2021.
Interest income for the nine months ended September 30, 2021, decreased by $49,000 versus the nine months ended September 30, 2020, due to a significant decrease in bank interest rates offset by an increase in bank balances resulting from funds raised in our Capital on DemandTM Sales Agreement with JonesTrading during the first quarter of 2021.
Liquidity and Capital Resources
Sources of Liquidity
We have incurred losses and cumulative negative cash flows from operations since our incorporation in December 2015 resulting in an accumulated deficit of approximately $38.6 million as of September 30, 2021. We anticipate that we will continue to incur losses for the foreseeable future. We expect that our R&D and G&A expenses will increase to enable the execution of our strategic plan. As a result, we anticipate that we will seek to raise additional capital within the next 12 months to fund our future operations. We will seek to obtain needed capital through a combination of equity offerings, debt financings, strategic collaborations and grant funding. To date, we have funded our operations through net proceeds from the initial public offering of our common stock, net proceeds from sales under our Capital on DemandTM Sales Agreement, private placements of our preferred and common stock, and the net receipt of funds related to the acquisition of camsirubicin. We anticipate that the currently available funds as of October 31, 2021, will fund our obligations through December 31, 2022.
We invest our cash equivalents in a money market account.
Cash Flows
The following table provides information regarding our cash flows for the nine months ended September 30, 2021, and 2020.
|
| Nine Months Ended September 30, |
|
| Nine Months Ended September 30, 2021, Compared to |
| ||||||
(in thousands) |
| 2021 |
|
| 2020 |
|
| Nine Months Ended September 30, 2020 |
| |||
Net cash used in operating activities |
| $ | (5,285 | ) |
| $ | (3,421 | ) |
| $ | (1,864 | ) |
Net cash provided by financing activities |
|
| 10,887 |
|
|
| 8,189 |
|
|
| 2,698 |
|
Effect of exchange rates |
|
| 3 |
|
|
| 1 |
|
|
| 2 |
|
Net increase in cash and cash equivalents |
| $ | 5,605 |
|
| $ | 4,769 |
|
| $ | 836 |
|
During the nine months ended September 30, 2021, we had net cash inflow of approximately $5,605,000, compared to net cash inflow of $4,769,000 during nine months ended September 30, 2020, an increase of approximately $836,000.
| 31 |
| Table of Contents |
Cash Flow Used in Operating Activities
The increase of approximately $1,864,000 in cash flow used in operating activities during the nine months ended September 30, 2021, compared to the nine months ended September 30, 2020, was a result of increased R&D and G&A cash operating expenses.
Cash Flow Used in Investing Activities
There was no cash flow used in investing activities for the nine months ended September 30, 2021, and 2020.
Cash Flow Provided by Financing Activities
The increase in cash flow provided by financing activities during the nine months ended September 30, 2021, compared to the nine months ended September 30, 2020, of approximately $2,698,000 was primarily due to the increase of net proceeds from sales of our common stock under our Capital on DemandTM Sales Agreement with JonesTrading.
Future Funding Requirements
To date, we have not generated any revenue from product sales. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate any revenue from product sales or royalties unless and until we obtain regulatory approval of and commercialize any of our current or future drug product candidates or we out-license or sell a drug product candidate to another party. At the same time, we expect our expenses to increase in connection with our ongoing development activities, particularly as we continue the research, development, future preclinical studies and clinical trials of, and seek regulatory approval for, our current and future drug product candidates. If we obtain regulatory approval of any of our current or future drug product candidates, we will need substantial additional funding for commercialization requirements and our continuing drug product development operations.
| 32 |
| Table of Contents |
As a company, we have not completed development through marketing approvals of any therapeutic products. We expect to continue to incur significant increases in expenses and increasing operating losses for the foreseeable future. We anticipate that our expenses will increase substantially as we:
· | advance the clinical development and execute the regulatory strategy for Validive; |
|
|
· | advance the clinical development and execute the regulatory strategy for camsirubicin; |
|
|
· | continue the preclinical activities and potentially enter clinical development of MNPR-101 and MNPR-101-derived radioimmunotherapeutics and companion diagnostics, to treat severe COVID-19 (patients with SARS-CoV-2 infection) and cancer; |
|
|
· | continue the preclinical activities, and potentially later-on enter clinical development, of MNPR-202 (and related analogs) for various cancer indications; |
|
|
· | acquire and/or license additional pipeline drug product candidates and pursue the future preclinical and/or clinical development of such drug product candidates; |
|
|
· | seek regulatory approvals for any of our current and future drug product candidates that successfully complete registration clinical trials; |
|
|
· | establish or purchase the services of a sales, marketing and distribution infrastructure to commercialize any products for which we obtain marketing approval; |
|
|
· | develop or contract for manufacturing/quality capabilities or establish a reliable, high quality supply chain sufficient to support our clinical requirements and to provide sufficient capacity to launch and supply the market for any product for which we obtain marketing approval; and |
|
|
· | add or contract for required operational, financial and management information systems and capabilities and other specialized expert personnel to support our drug product candidate development and planned commercialization efforts. |
We anticipate that the funds available as of October 31, 2021, will fund our obligations through December 2022. We have based this estimate on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development and commercialization of our drug product candidates, and the extent to which we enter into collaborations with third parties to participate in the development and commercialization of our drug product candidates, we are unable to accurately estimate with high reliability the amounts and timing required for increased capital outlays and operating expenditures associated with our current and anticipated drug product candidate development programs.
Our future capital requirements will depend on many factors, including:
· | the progress of clinical development and regulatory interactions and approvals of Validive; |
|
|
· | the progress of clinical development and regulatory interactions and approvals of camsirubicin; |
|
|
· | the progress of preclinical and clinical development of MNPR-101 and MNPR-101-derived radioimmunotherapeutics and companion diagnostics, to treat severe COVID-19 (patients with SARS-CoV-2 infection) and cancer, including activities through our collaboration with NorthStar; |
|
|
· | the progress of preclinical and potentially clinical development of MNPR-202 (and related analogs) |
|
|
· | the number and characteristics of other drug product candidates that we may license, acquire, invent or otherwise pursue; |
|
|
· | the scope, progress, timing, cost and results of research, preclinical development and clinical trials of future drug product candidates; |
|
|
· | the costs, timing and outcomes of seeking and obtaining and maintaining FDA and international regulatory approvals; |
| 33 |
| Table of Contents |
· | the costs associated with manufacturing/quality requirements and establishing or contracting for sales, marketing and distribution capabilities; |
|
|
· | our ability to maintain, expand and defend the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make in connection with the licensing, filing, defense and enforcement of any patents or other intellectual property rights; |
|
|
· | our need and ability to hire or contract for additional management, administrative, scientific, medical, sales and marketing, and manufacturing/quality and other specialized personnel or external expertise; |
|
|
· | the effect and timing of entry of competing products or new therapies that may limit market penetration or prevent the introduction of our drug product candidates or reduce the commercial potential of our product portfolio; |
|
|
· | our need to implement additional required internal systems and infrastructure; and |
|
|
· | the economic and other terms, timing and success of our existing collaboration and licensing arrangements and any collaboration, licensing or other arrangements into which we may enter in the future, including the timing of receipt of or payment to or from others of any milestone or royalty payments under these arrangements. |
Expenditures are expected to increase in the remainder of 2021 and onward for:
· | clinical research services and clinical site fees for our VOICE clinical program, including, if required, completing a second Phase 3 confirmatory clinical trial; |
|
|
· | process development, manufacturing costs, clinical trial expenses and clinical database management of camsirubicin in connection with the Phase 1b dose escalation clinical trial and other future clinical development; |
|
|
· | support of the development of MNPR-101-derived radioimmunotherapeutics and companion diagnostics to treat severe COVID-19 (patients with SARS-CoV-2 infection) and cancer, including activities through our collaboration with NorthStar; |
|
|
· | preclinical studies (and if successful, clinical studies) of MNPR-101, MNPR-202 (and related analogs); and |
|
|
· | employee compensation and consulting fees to support our product candidate programs including Validive, camsirubicin, MNPR-101, MNPR-101 RIT (uPRIT and related compounds) and companion diagnostics and MNPR-202 (and related analogs). |
We have activated clinical trial sites and are dosing patients in our VOICE clinical trial. In order to complete the VOICE clinical program, including, if required, completing a second Phase 3 confirmatory clinical trial, we will require additional funding in the millions or tens of millions of dollars (depending on if we have consummated a collaboration or partnership or neither for Validive), or find a suitable pharmaceutical partner, both of which we are planning to pursue within the next 12 months. There can be no assurance that any such events will occur. We have also initiated and commenced dosing in our Phase 1b camsirubicin clinical trial. We intend to continue evaluating drug product candidates for the purpose of growing our pipeline. Identifying and securing high-quality compounds usually takes time and related expenses; however, our spending could be significantly accelerated in the remainder of 2021 and onward if additional drug product candidates are acquired and enter clinical development. In this event, we may be required to expand our management team, and pay higher contract manufacturing costs, contract research organization fees, other clinical development costs and insurance costs that are not currently projected. The anticipated operating cost increases in the remainder of 2021 and onward are expected to be primarily driven by the funding of our VOICE clinical program, support of the camsirubicin clinical program, development of MNPR-101, MNPR-101 RIT (uPRIT and related compounds) and companion diagnostics and development of MNPR-202 (and related analogs). Beyond our need to raise additional funding in the coming 12 months to complete the VOICE clinical program, we will also need significant additional funding thereafter in order to commercialize Validive if approved for marketing, support of the camsirubicin clinical program beyond the Phase 1b clinical trial, to support our collaboration with NorthStar, further development of MNPR-101, MNPR-101 RIT (uPRIT and related compounds) and companion diagnostics, further development of MNPR-202 (and related analogs) and generally to support our current and any future product candidates through completion of clinical trials, approval processes and, if applicable, commercialization.
| 34 |
| Table of Contents |
Until we can generate a sufficient amount of product revenue to finance our cash requirements, we expect to finance our future cash needs primarily through a combination of equity offerings, debt financings, strategic collaborations and grant funding. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our current stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect our current stockholders’ rights. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with other parties, we likely will have to relinquish valuable rights to our technologies, future revenue streams, research programs or drug product candidates or grant licenses on terms that may not be favorable to us, which will reduce our future returns and affect our future operating flexibility. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our pipeline product development or commercialization efforts or grant rights to others to develop and market drug product candidates that we would otherwise prefer to develop and market ourselves.
Contractual Obligations and Commitments
License, Development and Collaboration Agreements
Onxeo S.A.
In June 2016, we executed an agreement with Onxeo S.A., a French public company, which gave us the exclusive option to license (on a world-wide exclusive basis) Validive (clonidine hydrochloride mucobuccal tablet; clonidine HCI MBT a mucoadhesive tablet of clonidine based on the Lauriad mucoadhesive technology). The agreement includes clinical, regulatory, developmental and sales milestones that could reach up to $108 million if we achieve all milestones, and escalating royalties from 5% to 10% on net sales. In September 2017, we exercised the option to license Validive from Onxeo for $1 million, but as of October 31, 2021, we have not been required to pay Onxeo any other funds under the agreement. We anticipate the need to raise significant funds to support the completion of clinical development and marketing approval of Validive.
Under the agreement, we are required to pay royalties to Onxeo on a product-by-product and country-by-country basis until the later of (1) the date when a given product is no longer within the scope of a patent claim in the country of sale or manufacture, (2) the expiry of any extended exclusivity period in the relevant country (such as orphan drug exclusivity, pediatric exclusivity, new chemical entity exclusivity, or other exclusivity granted beyond the expiry of the relevant patent), or (3) a specific time period after the first commercial sale of the product in such country. In most countries, including the U.S., the patent term is generally 20 years from the earliest claimed filing date of a non-provisional patent application in the applicable country, not taking into consideration any potential patent term adjustment that may be filed in the future or any regulatory extensions that may be obtained. The royalty termination provision pursuant to (3) described above is shorter than 20 years and is the least likely cause of termination of royalty payments.
The Onxeo license agreement does not have a pre-determined term, but expires on a product-by-product and country-by-country basis; that is, the agreement expires with respect to a given product in a given country whenever our royalty payment obligations with respect to such product have expired. The agreement may also be terminated early for cause if either we or Onxeo materially breach the agreement, or if either we or Onxeo become insolvent. We may also choose to terminate the agreement, either in its entirety or as to a certain product and a certain country, by providing Onxeo with advance notice.
Grupo Español de Investigación en Sarcomas (“GEIS”)
In June 2019, we executed a clinical collaboration with GEIS for the development of camsirubicin in patients with advanced soft tissue sarcoma (“ASTS”). Following completion of the Phase 1b clinical trial in the U.S. that we initiated in the third quarter of 2021 with the first patient dosed in October 2021, we continue to expect that GEIS will sponsor and lead a multi-country, randomized, open-label Phase 2 clinical trial to evaluate camsirubicin head-to-head against doxorubicin, the current first-line treatment for ASTS. We will provide study drug and supplemental financial support for the clinical trial estimated to average approximately $2 million to $3 million per year. During the three and nine months ended September 30, 2021, in preparation for the Phase 1b clinical trial, we incurred $0.3 million and $1.0 million, respectively, in expenses including clinical material manufacturing-related costs and database management expenses. During the three and nine months ended September 30, 2020, we incurred $0.4 million and $0.6 million, respectively, in expenses under the GEIS agreement and other clinical-related expenses including clinical material manufacturing-related costs and database management expenses. We can terminate the agreement by providing GEIS with advance notice, and without affecting our rights and ownership to any intellectual property or clinical data.
XOMA Ltd.
Pursuant to a non-exclusive license agreement with XOMA Ltd. for the humanization technology used in the development of MNPR-101, we are obligated to pay XOMA Ltd. clinical, regulatory and sales milestones which could reach up to $14.925 million if we achieve all milestones for MNPR-101. The agreement does not require the payment of sales royalties. There can be no assurance that we will achieve any milestones. As of October 31, 2021, we had not reached any milestones and had not been required to pay XOMA Ltd. any funds under this license agreement.
| 35 |
| Table of Contents |
Service Providers
In the normal course of business, we contract with service providers to assist in the performance of R&D, including drug product manufacturing, process development, clinical development, and G&A including financial strategy, audit, tax and legal support. We can elect to discontinue the work under these agreements at any time. We could also enter into collaborative research and development, contract research, manufacturing and supplier agreements in the future, which may require upfront payments and/or long-term commitments of cash.
Office Lease
We are currently leasing office space in the Village of Wilmette, Illinois for $4,487 per month on a month-to-month basis, and we anticipate that we will lease additional space in the future as we hire additional personnel.
Legal Contingencies
We are currently not, and to date have never been, a party to any adverse material legal proceedings.
Indemnification
In the normal course of business, we enter into contracts and agreements that contain a variety of representations and warranties and provide for general indemnification. Our exposure under these agreements is unknown because it involves claims that may be made against us in the future, but that have not yet been made. To date, we have not paid any claims or been required to defend any action related to our indemnification obligations. However, we may record charges in the future as a result of these indemnification obligations.
In accordance with our Second Amended and Restated Certificate of Incorporation, Amended and Restated Bylaws and the indemnification agreements entered into with each officer and non-employee director, we have indemnification obligations to our officers and non-employee directors for certain events or occurrences, subject to certain limits, while they are serving at our request in such capacity. There have been no claims to date.
Off-Balance Sheet Arrangements
To date, we have not had any off-balance sheet arrangements, as defined under the SEC rules.
Item 4. Controls and Procedures
Our Chief Executive Officer and Chief Financial Officer have provided certifications filed as Exhibits 31.1 and 32.1, and 31.2, respectively. Such certifications should be read in conjunction with the information contained in this Item 4 for a more complete understanding of the matters covered by those certifications.
(a) Disclosure Controls and Procedures
We carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of our disclosure controls and procedures as of September 30, 2021, pursuant to Rules 13a15(e) and 15d15(e) under the Exchange Act. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures, as of such date, were effective.
(b) Changes in Internal Control over Financial Reporting
We have concluded that the condensed consolidated financial statements and other financial information included in this Quarterly Report on Form 10-Q fairly present in all material respects our financial condition, results of operations and comprehensive loss and cash flows as of, and for, the periods presented.
There have been no changes in our internal control over financial reporting during the three months ended September 30, 2021, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| 36 |
| Table of Contents |
PART II. OTHER INFORMATION
Item 1A. Risk Factors
There have been no material changes in information regarding our risk factors as described in Item 1A of our Annual Report on Form 10-K as filed with the SEC on March 25, 2021.
Item 6. Exhibits
The following exhibits are filed as part of this Quarterly Report on Form 10-Q.
Exhibit |
| Document |
| Incorporated by Reference From: |
| Certification of Chandler D. Robinson, Chief Executive Officer |
| Filed herewith | |
|
| Filed herewith | ||
|
| Filed herewith | ||
101.INS |
| Inline XBRL Instance Document |
|
|
101.SCH |
| Inline XBRL Taxonomy Extension Schema |
|
|
101.CAL |
| Inline XBRL Taxonomy Extension Calculation Linkbase |
|
|
101.DEF |
| Inline XBRL Taxonomy Extension Definition Linkbase |
|
|
101.LAB |
| Inline XBRL Taxonomy Extension Label Linkbase |
|
|
101.PRE |
| Inline XBRL Taxonomy Extension Presentation Linkbase |
|
|
104 |
| Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) |
|
|
| 37 |
| Table of Contents |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
MONOPAR THERAPEUTICS INC. | |||
|
| ||
Dated: November 11, 2021 | By: | /s/ Chandler D. Robinson | |
Name: Chandler D. Robinson | |||
Title: Chief Executive Officer and Director (Principal Executive Officer) | |||
MONOPAR THERAPEUTICS INC. | |||
|
| ||
Dated: November 11, 2021 | By: | /s/ Kim R. Tsuchimoto | |
Name: Kim R. Tsuchimoto | |||
Title: Chief Financial Officer (Principal Financial Officer) | |||
| 38 |